- +1
【复材资讯】明军/李茜电解液锂金属篇:电解液组分与锂金属沉积的具象化

【研究背景】
电解液研究自2018年以来在金属(离子)电池领域再次引起广泛关注,这不仅是因为改变电解液组分能够显著提升电池性能,更是因为研究者发现电解液组分的变化,能够改变电解液溶剂化结构及去溶剂化行为,进而影响电极/电池性能。该系列研究明晰了其影响过程,丰富了对传统电极表面形成的稳定固体电解质界面膜(SEI)稳定电池性能的理解,将电解液领域的研究重点从SEI膜转移至电解液溶剂化结构,促进了研究者对金属离子(脱)嵌入化学的深入思考,即金属离子(M+)与溶剂之间的相互作用决定了(脱)嵌入过程的可逆性。基于此,研究进一步发现,成膜添加剂,例如碳酸丙烯酯(PC)基电解液中的硫酸乙烯酯(DTD)以及锂硫电池中的硝酸锂(LiNO3)液也能显著改变电解液溶剂化结构,从而影响电极性能。目前,锂离子电池(LIBs)中石墨电极的性能可以通过锂离子(Li+)与溶剂的相互作用解释,但对于下一代锂金属电池中锂沉积/剥离行为的理解仍值得深入研究。尤其,区分电解液溶剂化、去溶剂化和SEI膜形成的影响对于在分子水平上理解电解液行为与电极性能至关重要。
醚基电解液作为锂金属电池中最常用的电解液,通过调控溶剂种类(乙二醇二甲醚(DME)、DME/1, 3-二氧五环(DOL))、比较不同锂盐的作用(双氟磺酰亚胺锂盐(LiFSI)、双三氟甲烷磺酰亚胺锂(LiTFSI))、以及引入LiNO3添加剂,能够显著影响电解液溶剂化结构,进而调控锂沉积/剥离行为。尽管含不同锂盐和不同浓度的DME/DOL基电解液,包括高浓度电解液已被广泛研究,然而对于溶剂、盐、以及添加剂如何在分子尺度影响锂沉积行为仍缺乏研究。目前,常见的解释多归因于形成稳定SEI膜以提高电极性能,尤其是溶剂化结构诱导的阴离子主导的SEI膜。如何系统描述溶剂(如DME、DOL)、锂盐(如LiTFSI、LiFSI)和添加剂(如LiNO3)在不同电解液中的不同作用,以及这些因素如何在分子尺度影响电极和电池性能仍然缺乏详细的研究。因此,厘清电解液中溶剂、离子行为,并建立其与界面去溶剂化行为和锂沉积/剥离化学之间的关系,将会对研究下一代锂金属、锂硫和锂空气电池,实现超过500wh kg-1能量密度的电池体系构建产生重要的影响。尤其,理解电解液行为与锂沉积/剥离过程,尤其是分子尺度上离子、溶剂的相互作用,对缓解枝晶的形成、提高电池安全性至关重要。
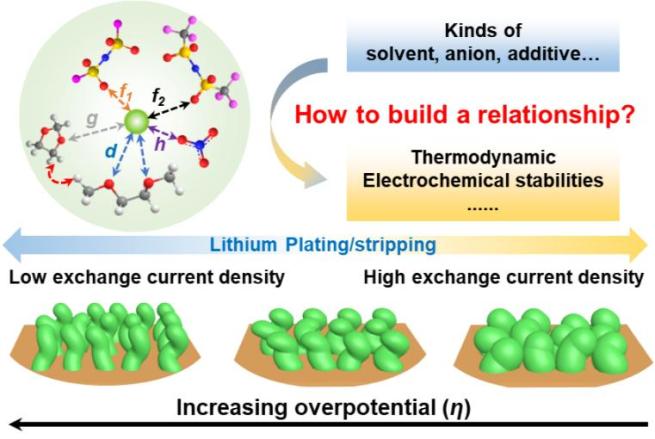
图1. 离子、溶剂相互作用与锂沉积/剥离动力学及热力学行为研究
【研究介绍】
近日中科院长春应化所明军研究员、马征、谢宏亮特别研究助理系统地探究了电解液溶剂化结构、锂沉积形态、以及循环效率之间的关系,以确定控制锂沉积/剥离化学的根本因素。研究发现,通过特定的离子、溶剂配置能够调控Li+溶剂化结构,促进电解液/电极界面处的Li+去溶剂化过程,提高交换电流密度,降低锂沉积过程中的成核过电位,实现锂金属的均匀沉积及高的库伦效率。此外,弱Li+-溶剂配位簇有助于Li+迁移动力学和去溶剂化行为,促进均匀的锂成核和生长,同时增强电解液的稳定性。尤其,本研究阐明了每种电解液成分如何在分子水平上影响Li+溶剂化结构及界面去溶剂化动力学及热力学稳定性,并提出了一个与不同电解液成分相关的锂沉积/剥离过程的界面模型,以阐明电解液中离子、溶剂配置及相互作用对锂沉积/剥离化学的影响。这些发现解决了长期以来调控锂枝晶生长的难题,并为设计高性能锂金属电池电解液提供了指导方针,并以“Electrolyte Solvent-Ion Configuration Deciphering Lithium Plating/Stripping Chemistry for High-Performance Lithium Metal Battery”为题,发表在国际知名学术期刊Advanced Functional Materials上,中科院长春应化所的副研究员李茜为本文第一作者。
【内容表述】
1. 锂沉积/剥离行为分析
本研究以常用的醚基电解液(即DME/DOL)为例,旨在开发一种分子级模型,建立电解液微观结构与锂沉积/剥离行为之间的关系,以阐明电解液中各组分的作用。首先,使用Li||Li对称电池和Li||Cu不对称电池研究了电解液组分对锂金属负极的兼容性和循环稳定性的影响(图1)。具体而言,使用DME作为电解液中的唯一溶剂来溶解锂盐,形成DME基电解液。然后逐步将DOL引入上述电解液中,最后添加LiNO3作为电解液添加剂。此外,还分别评估了LiTFSI和LiFSI盐对电极性能的影响,以平行比较锂盐引起的电解液性能差异。这种方法能够研究随着电解液成分的逐渐改变锂沉积/剥离行为的变化,从而揭示电解液成分与锂沉积/剥离过程之间的关系,并阐明各成分的作用。首先,使用LiTFSI/DME基电解液的Li||Li对称电池考察锂沉积/剥离过程的差异,其中电解液浓度、电流密度和截止容量分别控制在 1.0 M、0.5 mA cm-2和1 mAh cm-2。具体而言,Li||Li对称电池的初始极化电压高达30-40 mV,电池寿命不足80 h,表明LiTFSI/DME基电解液与锂金属的不稳定性及不兼容性。随后,向LiTFSI/DME基电解液中加入DOL后,极化电压降至约20 mV,电池变得更为稳定。最后加入LiNO3,极化电压进一步降低(15 mV),并稳定循环200 h。这些结果表明,DOL和LiNO3均能显著降低电池极化,同时增强电解液与锂金属的兼容性。相比之下,在LiFSI基电解液的平行实验中,获得相同的趋势,且基于LiFSI基电解液的Li||Li对称电池表现出更低的极化电压和更好的循环稳定性,表明LiFSI在促进锂沉积/剥离过程方面具有优势。上述结果表明,TFSI-和FSI-阴离子的不同性质、溶剂的类型、以及有无添加剂均会导致锂沉积/剥离行为的差异,这反过来又会影响电解液与锂金属负极的兼容性,并最终决定电池的使用寿命。此外,使用Li||Cu不对称电池进一步研究了各种电解液成分对库仑效率和循环稳定性的影响,结果呈现出相同的趋势。基于此,无论Li||Li对称电池还是Li||Cu不对称电池的电化学性能,均表明电解液成分显著影响锂镀/剥离化学的可逆性以及循环过程中电解液的稳定性。
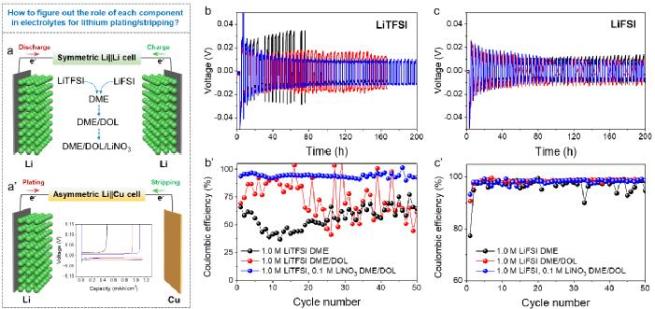
图2. 电池模型构建及锂沉积/剥离行为研究
2. 锂沉积界面动力学分析
首先,通过锂沉积动力学进一步评估电解液成分及其差异对锂沉积/剥离化学的影响(图2)。在Li||Li对称电池中测量的交换电流密度以及在Li||Cu不对称电池中测试的成核过电位有效地检测到了电解液成分变化所产生的影响。当在LiTFSI/DME基电解液中加入DOL和LiNO3时,Li||Li对称电池的交换电流密度从初始的5.5 mA cm-2增加到12.1 mA cm-2和16.7 mA cm-2。由于交换电流密度(J0)与恒定电流密度下的过电位(η)成反比,因此在交换电流密度更大的情况下,预期会有一个更低的成核过电位(ηn),从而促进均匀的锂沉积。这一点在Li||Cu不对称电池中得到了进一步的证实,即添加DOL和LiNO3时,在锂沉积/剥离过程成核过电位逐步降低,从77 mV逐步降至68 mV及53 mV。这些结果表明DOL和LiNO3均可加快电解液/电极界面处的反应速率,并促进锂沉积/剥离过程。此外,电解液/电极界面处锂离子去溶剂化的活化能进一步证实了上述观点。当向LiTFSI/DME基电解液中加入DOL和LiNO3时,去溶剂化能从55.95 kJ mol-1下降到45.14 kJ mol-1和42.31 kJ mol-1。这些发现表明,通过在电解液中添加DOL和LiNO3,可以调节Li+溶剂化结构,并降低锂离子去溶剂化的活化能,从而显著提高锂沉积过程中的电荷转移动力学(即Li+去溶剂化动力学)。同时,在LiFSI/DME基电解液中也观察到了类似的趋势,进一步证明了电解液成分的重要性。其次,通过锂成核尺寸的变化进一步解析了电解液中各成分所起到的作用,也为交换电流密度和成核过电位的讨论提供了支撑。根据经典的成核理论,锂沉积过程中的成核尺寸与过电位成反比。也就是说,较低的成核过电位促进较大晶核的形成,从而确保电流均匀分布,减少电场畸变,进而促进均匀的锂沉积,以防止孔隙形成和枝晶生长。在不同电解液中沉积的锂的形态不仅验证了这种关系,还突出了每种电解液组分的作用。例如,DME基电解液中逐步加入DOL和LiNO3,沉积在铜箔上的锂逐渐增大,沉积形态变得更加平滑,与前面的分析结果一致。这些趋势在基于LiTFSI和LiFSI的电解液中均十分明显,突显了DOL和LiNO3在促进锂沉积/剥离过程以及提高电解液与锂金属负极兼容性方面所起的关键作用。因此,优化电解液成分以促进Li+去溶剂化、增加交换电流密度和降低成核过电势,是实现均匀锂沉积的有效策略。
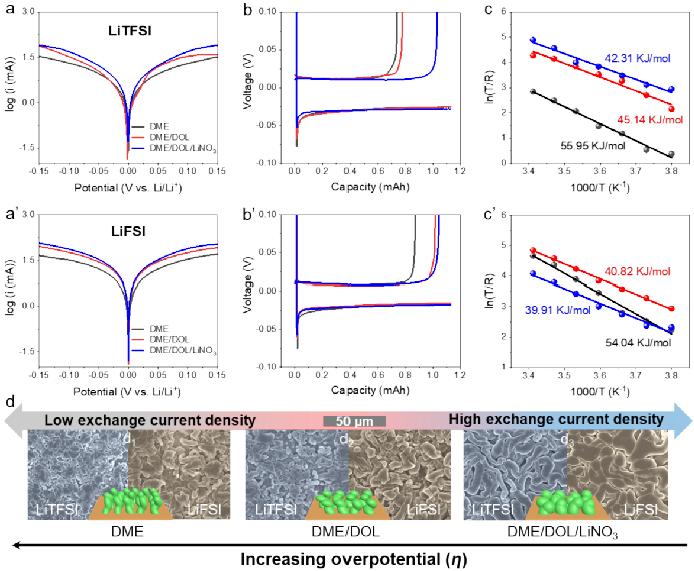
图3. 不同电解液中的锂沉积界面动力学行为研究
3. 电解液溶剂化结构表征
不同电解液之间锂金属沉积行为的差异与Li+去溶剂化过程密切相关。首先,利用核磁共振(NMR)光谱研究了每个电解液成分引起的微观结构变化(图4a-c)。7Li NMR光谱显示,在LiTFSI/DME基电解液中加入DOL、以LiFSI替代LiTFSI、以及添加LiNO3添加剂时出现蓝移(即向更高值移动)。这种蓝移表明阴离子或溶剂对锂离子的屏蔽效应减弱,意味着锂离子与溶剂的相互作用变弱,有助于Li+去溶剂化过程。同时,DME中C-O-C键的17O NMR信号产生红移(即向更低的化学位移值移动),表明17O核周围电子云密度的增加,与7Li NMR结果一致。Li+与DME之间的相互作用减弱,有助于锂离子在锂金属负极界面去溶剂化,从而促进锂的均匀沉积,同时降低过电位,提高库仑效率。值得注意的是,DME/DOL基电解液中DME的1H NMR光谱相较于纯DME基电解液中的光谱向高场偏移,表明DME中H原子周围的电子密度增加,这可能是由于DOL的C-O-C键中的富电子氧(δ-O)与DME中-CH2和-CH3中的缺电子氢(δ+H)之间的偶极-偶极相互作用所致。这种相互作用可能削弱DME与Li+的相互作用,与上述核磁共振分析结果一致。
此外,通过拉曼光谱进一步解析了溶剂、盐和添加剂对Li+溶剂化结构的影响(图4d)。在LiTFSI/DME基电解液中,739 cm-1和744 cm-1处的峰分别对应于溶剂分离离子对(SSIP)和接触离子对(CIP)。随着DOL的加入,SSIP峰面积从82%降至78%,而CIP峰面积从18%增至22%,证实了Li+溶剂化结构的变化。这种转变可能归因于DME-DME到DME-DOL溶剂-溶剂相互作用的改变,其中DME与DOL之间的相互作用使得TFSI-更容易靠近Li+,从而形成更多的CIPs。当使用LiFSI代替LiTFSI时,峰位置(即720 cm-1)有明显的位移,这可能归因于TFSI-被FSI-替代,对应于FSI-的S-N-S拉伸振动的变化。在LiFSI/DME/DOL基电解液中出现了745 cm-1的阳离子-阴离子聚集峰(AGG),且717 cm-1处的SSIP峰面积从78%减少至52%,而727 cm-1处的CIP峰面积则从22%增加至43%,表明FSI-与Li+之间的相互作用强于TFSI-与Li+之间的相互作用。进一步在LiFSI/DME/DOL基电解液中添加LiNO3后,SSIP峰面积从52%减少至47%,而CIP和AGG峰面积分别从43%增加至47%和从5%增加至6%,表明FSI-与Li+之间的相互作用变得更强,进一步削弱了Li+-溶剂相互作用,促进Li+去溶剂化,解释了LiFSI/DME/DOL/LiNO3基电解液具有出色电化学性能的原因。其次,径向分布函数(RDF)的进一步支撑了上述光谱分析的结果,从理论计算的角度证明了上述结论的有效性。(图4e-h)。
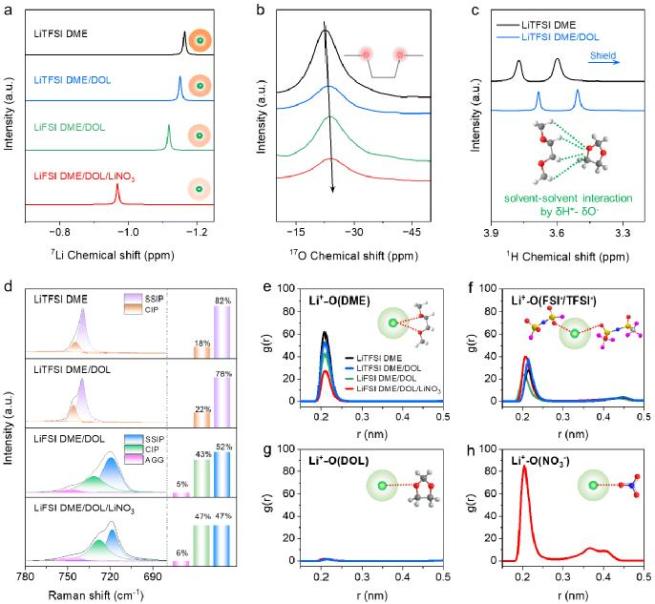
图4. 不同电解液溶剂化结构表征
作者利用NOESY进一步证实了DME/DOL基电解液中存在溶剂-溶剂相互作用,并阐明了DOL和LiNO3的作用、以及LiTFSI和LiFSI之间的差异在电解液组分中所起到的作用(图5)。当在LiTFSI/DME基电解液中添加DOL时,检测到DME-H和DOL-H之间存在微弱的交叉弛豫现象(即由DME中位置①的H原子和DOL中位置②的H原子引起),表明DME和DOL之间存在弱溶剂-溶剂相互作用。当使用LiFSI取代LiTFSI和引入LiNO3添加剂时,DME和DOL之间的相互作用逐渐增强,与一维光谱数据相吻合。上述光谱结果表明溶剂-溶剂相互作用、阴离子类型、以及添加剂能够显著影响溶剂化结构及界面去溶剂化行为,从而实现均匀的锂沉积及较高的锂沉积/剥离可逆性。基于上述分析,Li⁺第一层溶剂化层的变化主要受Li⁺与DME/阴离子之间的相互作用以及DME与DOL之间的分子间相互作用的影响。随着DOL加入、LiTFSI被LiFSI替代、以及LiNO3的引入,Li⁺-DME的配位作用逐渐减弱(即d1< d2 < d3 < d4),而Li⁺-阴离子相互作用逐渐增强(即f1 > f2 > f3 > f4)。这些趋势与7Li NMR低场位移和17O NMR的高场位移以及拉曼光谱中CIP和AGG的峰比增加一致。基于此,确定了包括溶剂类型、盐种类以及添加剂在内的每个成分如何通过溶剂和阴离子的重新配置来影响Li+溶剂化结构及界面去溶剂化团簇的动力学及热力学稳定性,特别是对Li+-溶剂相互作用的影响,这些相互作用反过来又能决定锂沉积/剥离行为以及沉积锂的形态。
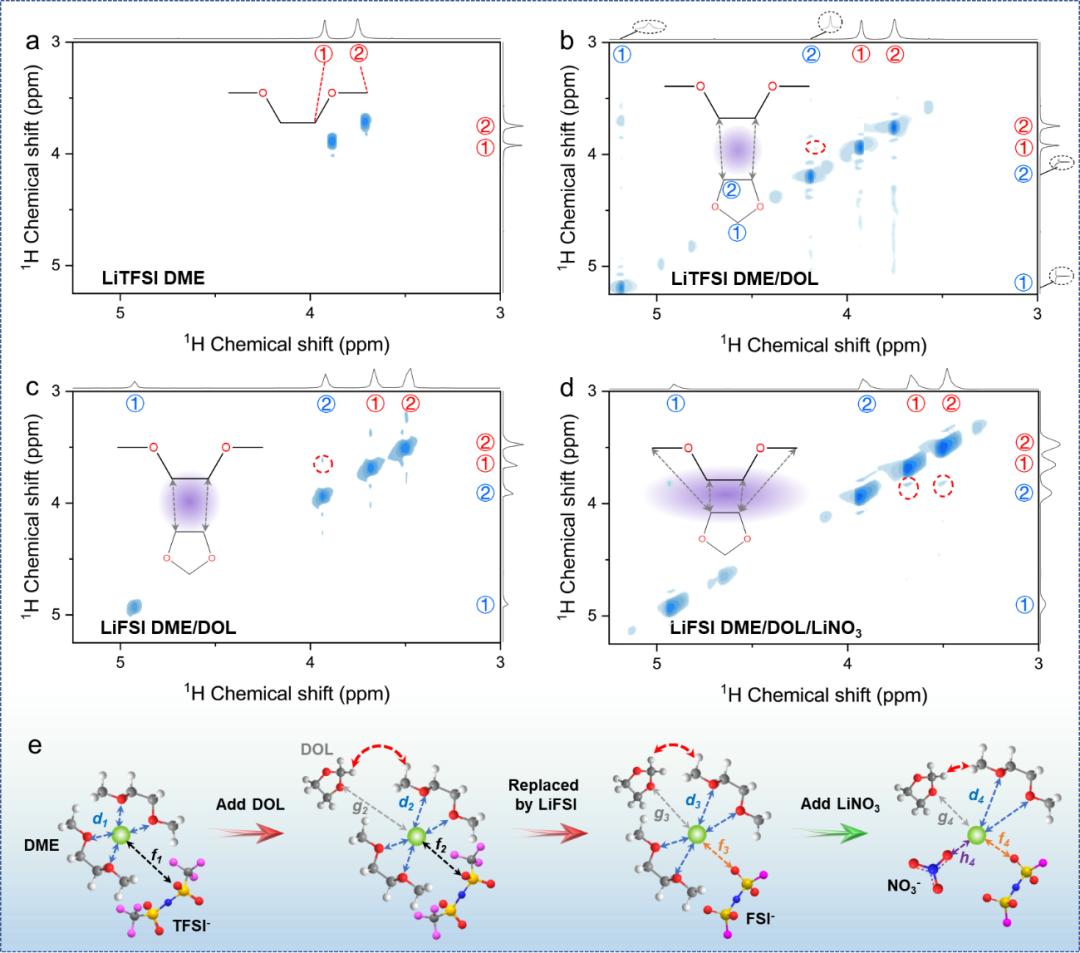
图5. 分子间相互作用表征和相互作用示意图
4. 电解液溶剂化结构及界面模型构建
基于上述分析,作者提出Li+溶剂化结构(Li+[溶剂]x[阴离子],其中x是溶剂与Li+的摩尔比)和相应的界面模型,在分子尺度上建立Li+溶剂化结构、界面去溶剂化行为与锂沉积/剥离过程之间的关系(图6)。在LiTFSI/DME基电解液中(Li+[DME]9.62[TFSI-]),由于DME与Li+的结合能力很强(即双齿螯合效应),Li+-DME作用(即d1)很强,导致TFSI-与溶剂化的Li+之间的静电相互作用很弱(即f1)。在电极界面处Li+沉积过程中,静电排斥作用使得TFSI-远离锂金属界面(即f1'),导致Li+-DME形成的离子簇占据主导地位,减缓了Li+去溶剂化动力学。其次,由于Li+-DME的强相互作用(即d1'),DME极化严重,导致DME的还原分解严重。同时,Li+-DME与锂金属界面紧密接触(即u1'),容易被锂金属负极表面的电子攻击,进一步加剧电解液分解,从而影响锂成核过电位和沉积形态,最终降低电池效率。
将DOL添加到LiTFSI/DME基电解液中,溶剂化结构转变为Li+[DME]4.81[DOL]7.19[TFSI-]。其中,DOL与DME之间的分子间相互作用减少了DME与Li+之间的相互作用(即d2,d2 > d1),同时增强TFSI-与Li+之间的相互作用(即f2,f2 < f1)。同时,TFSI-的负电荷能够中和Li+的正电荷,进一步削弱Li+-DME相互作用。因此,在电极界面处Li+更容易从Li+-DME团簇中脱出(即d2',d2' > d1'),从而降低成核过电位和界面阻抗,进而提高电池性能。此外,DME与DOL之间的分子间相互作用可以通过电子离域降低DME的极化,并且增强TFSI-与Li+之间的相互作用(即f2',f2' < f1'),使Li+-DME/DOL团簇稍微远离锂金属负极(即u2',u2' > u1'),不易受到锂金属负极上电子的攻击,从而增强电解液电化学稳定性。
电解液中LiTFSI和LiFSI之间的差异也可以通过Li+溶剂化结构和界面模型以图形方式进行描述。例如,在1.0 M LiFSI DME/DOL电解液中,Li+溶剂化结构可以表示为Li+[DME]4.81[DOL]7.19[FSI-]。在这个结构中,Li+与DME之间的相互作用进一步减弱(即d3,其中d3 > d2 > d1),而Li+与FSI-(即f3,其中f3 < f2 < f1)之间的相互作用增强。这是由于DOL和DME之间更强的偶极-偶极相互作用以及FSI-的较低空间阻碍和更集中电子云所致。因此,在锂沉积过程中,由于Li+与DME溶剂之间的相互作用减弱(即d3',其中d3' > d2' > d1'),Li+更容易脱溶剂化并沉积。此外,FSI-的负电荷有助于中和溶剂化结构和界面模型中的Li+的正电荷(即f3',其中f3' < f2' < f1'),从而降低Li+-DME/DOL复合物与锂金属负极之间的库仑相互作用,使Li+-DME/DOL复合物远离电极表面(即u3',其中u3' > u2' > u1'),使其更不容易受到锂金属上电子的攻击。因此,用LiFSI替代LiTFSI有助于在锂金属负极界面实现更有效的Li+去溶剂化和均匀的锂沉积,从而提高电解液与锂金属负极的兼容性。
进一步在上述电解液中引入LiNO3添加剂(即Li⁺[DME]4.37[DOL]6.53[FSI-]0.91[NO3-]0.09),NO3-阴离子竞争进入第一层溶剂化壳层(即h4),削弱Li⁺-DME的配位作用(即d4,其中d4 > d3 > d2 > d1),同时增强Li⁺-FSI-的相互作用(即f4,其中f4 < f3 < f2 < f1)。在Li⁺去溶剂化过程中,Li⁺周围的NO3-(即h4')和FSI-(即f4',其中f4' < f3' < f2' < f1')能够显著削弱Li⁺-DME的相互作用(即d4',其中d4' > d3' > d2' > d1'),从而促进锂沉积动力学和形成更均匀的锂沉积形态。此外,由于Li+被NO3-和FSI-中和后电荷相互作用减弱,使得Li+-DME/DOL复合物距离锂金属负极更远(即u4',其中u4' > u3' > u2' > u1'),削弱了锂金属负极上电子对电解液的攻击,从而增强电解液的电化学稳定性,显著提高了其与锂金属的兼容性。
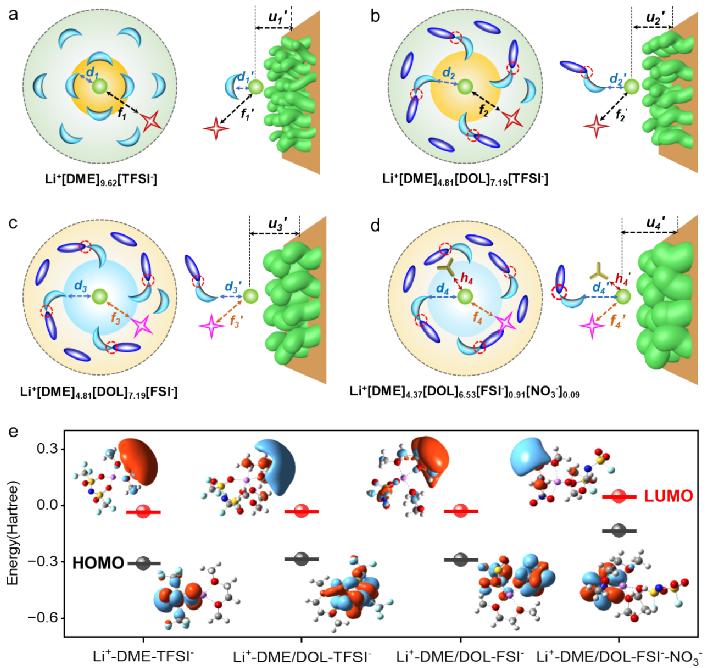
图6. 电解液溶剂化结构及界面模型构建
此外,作者还通过Li+-溶剂-阴离子配合物轨道之间的能量差(ΔE = HOMO'-LUMO)以评估电极界面处去溶剂化团簇的电化学稳定性(即电解液稳定性)。能量差(ΔE)越大表明界面上不同分子轨道之间的电子转移越困难,去溶剂化团簇的稳定性较高。结果表明,Li+-DME-TFSI-团簇的LUMO能级水平(-0.0317 Hartree)低于Li+-DME/DOL-TFSI-团簇(-0.0289 Hartree),表明DOL的存在削弱了Li+-溶剂相互作用,有效地提高了去溶剂化团簇的还原稳定性,并增强了电解液的热力学性质。当TFSI-被FSI-取代和引入LiNO3添加剂后,去溶剂化团簇的LUMO能级显著提高,表明电解液的抗还原能力逐渐增强,与Li||Cu不对称电池的电化学性能一致。总而言之,通过调控电解液中溶剂化结构,不仅在动力学角度降低了成核过电位、增加交换电流密度,以降低电池极化,还能在热力学角度抑制电解液分解,提高电池循环效率。
5. 全电池验证
DOL和LiNO3的作用以及LiFSI相对于LiTFSI的优势在Cu@Li||LiFePO4全电池中得到了进一步的验证(图7)。首先,在LiTFSI/DME基电解液中添加DOL和LiNO3后,初始库仑效率从93.6%(164 mAh g-1/154 mAh g-1)提高到95.6%(164 mAh g-1/158 mAh g-1)和97.1%(163 mAh g-1/158 mAh g-1)。经过50次循环后,容量保持率从0.14%提高到5.1%和95.7%。当使用LiFSI代替LiTFSI,观察到类似的趋势。上述Cu@Li||LiFePO4全电池循环性能差异进一步证实了电解质溶剂化结构及界面去溶剂化行为调控对于电池性能的影响。此外,使用LiFSI基电解液的Cu@Li||LiFePO4电池性能优于LiTFSI基电解液的电池性能,这一结果与Li||Li对称电池和Li||Cu不对称电池中测试结果一致,再次证明了LiFSI在稳定电解液和提升电池性能方面的优势。上述结果表明,在全电池体系中每个电解液成分的影响都能得到很好的保留。此外,LiFSI/DME/DOL/LiNO3基电解液体系,表现出较好的倍率性能,在0.2C、1C、2C、3C、4C和5C的电流密度下,分别实现了159、145、130、114、102和88 mAh g-1的高比容量,进一步证明了改性电解质的稳定性和可靠性。更重要地,通过优化电极材料(例如颗粒尺寸、表面处理工艺)、电极制备工艺(例如浆料比例、负载量)以及工步条件(例如配置、截止电压等)能够进一步改善电池性能。
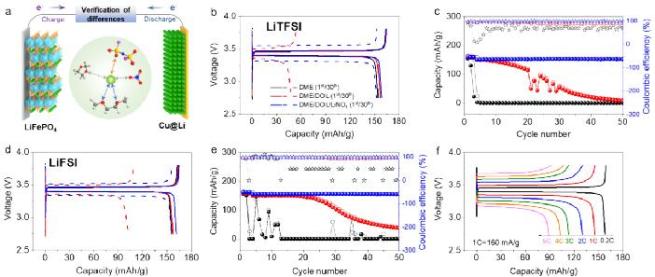
图7. Cu@Li ||LiFePO4全电池性能
【结论】
该研究构建了一个分子层面的电解液溶剂化结构和界面模型,以阐明电解液中各成分的作用,旨在阐明影响锂金属电池性能变化的关键因素。通过在分子尺度上建立锂沉积/剥离行为与电解液成分之间的关系,辨识了常用的DME基电解液中DOL溶剂、LiNO3添加剂、以及不同盐种类的作用,揭示了DME-DOL分子间相互作用、阴离子构型、以及NO3-的强配位能力在锂沉积过程中对电极界面溶剂化行为动力学及热力学稳定的影响。该研究以锂硫电池和锂金属电池中常用的醚基电解液为例,阐明了溶剂、盐和添加剂如何在分子层面影响电解液行为,进而影响锂沉积形态及锂沉积/剥离化学行为,体现了溶剂化结构及分子界面模型的普遍性。该研究为调节锂沉积/剥离行为的电解液工程提供了不同的视角,有助于开发稳定的锂金属负极,并推动锂金属电池领域的发展。
原文链接:https://doi.org/10.1002/adfm.202420327
免责声明:中国复合材料学会微信公众号发布的文章,仅用于复合材料专业知识和市场资讯的交流与分享,不用于任何商业目的。任何个人或组织若对文章版权或其内容的真实性、准确性存有疑议,请第一时间联系我们。我们将及时进行处理。
继续滑动看下一个轻触阅读原文

中国复合材料学会向上滑动看下一个
原标题:《【复材资讯】明军/李茜电解液锂金属篇:电解液组分与锂金属沉积的具象化》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。


- 一键“入夏”
- 中日经济高层对话达成二十项共识
- 大陆打出“惩独”组合拳备受关注
- 北京市长殷勇会见英国阿斯利康公司全球首席执行官
- 哪吒系列电影总票房破200亿元
- 网络流行词,指避免陷入无意义的“内卷式”竞争
- 植物细胞壁的主要成分


- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2025 上海东方报业有限公司

