- +1
【科技前沿】Protein & Cell:甘波谊团队全面分析SLC7A11/xCT在癌症中的作用—…
半胱氨酸是一种非常重要的氨基酸,在蛋白质合成、翻译后修饰和氧化还原维持中具有多种作用。半胱氨酸是谷胱甘肽的限速前体,谷胱甘肽是一种由三个氨基酸(半胱氨酸、谷氨酸和甘氨酸)组成的三肽,是细胞内最丰富的抗氧化剂。胞内半胱氨酸可通过从头生物合成(通过转硫途径)或通过蛋白质降解回收,大多数癌细胞主要依靠胱氨酸转运蛋白系统 Xc-(由催化亚基SLC7A11和伴侣亚基SLC3A2组成)从细胞外环境获得胱氨酸,然后通过消耗NADPH的还原反应在胞质中将其转化为半胱氨酸;半胱氨酸随后被用于合成谷胱甘肽(以及其他生物分子)。越来越多的研究表明,SLC7A11介导的胱氨酸摄取在抑制氧化反应和维持氧化应激条件下的细胞存活中发挥关键作用。
近年来,作为一种新的细胞死亡机制,铁死亡引起了科学界的极大兴趣。常见的癌症疗法,例如免疫疗法和放射疗法,可以部分通过调节SLC7A11的表达来诱发铁死亡。但是,SLC7A11的表达及其转运蛋白活性的多种调控,以及其如何精细控制癌细胞的铁死亡仍然充满未知,并激起了科学界的广泛关注。此外,另一个有趣的方向是SLC7A11如何诱导癌细胞的营养依赖性。
近日,美国MD安德森癌症中心甘波谊研究组在Protein & Cell杂志上发表了名为Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy的综述,总结了SLC7A11表达及其转运蛋白活性的多种调控机制,并讨论了SLC7A11如何通过抑制铁死亡以及其他铁死亡非依赖性的功能促进肿瘤的发展。最后,作者还讨论了SLC7A11诱导癌细胞营养依赖性的代谢基础。

1
SLC7A11的调控机制
为确保SLC7A11在维持氧化还原稳态中的适当功能,细胞通过多种机制对SLC7A11的表达和活性进行严格的调控。作者从转录因子和表观遗传调控因子的转录调控,以及转录后调控机制(对mRNA水平的调控)、蛋白质稳定性、,亚细胞定位和转运蛋白活性等方面展开了对SLC7A11调控机制的深入讨论(图1)。
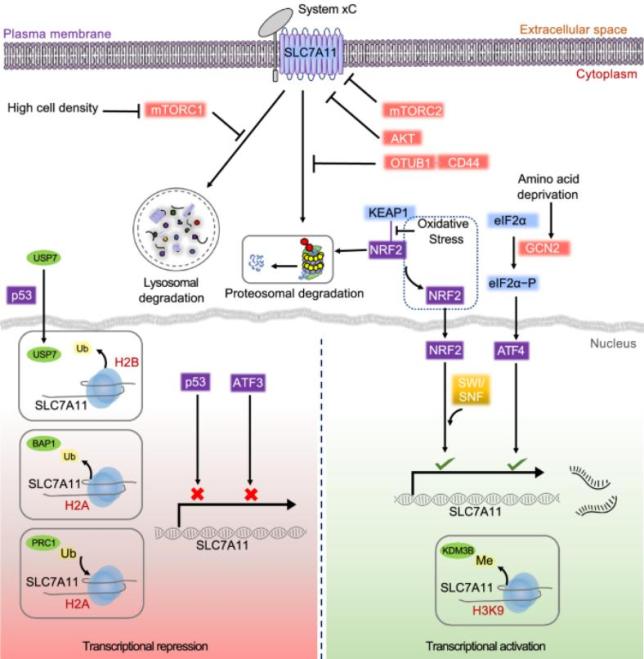
图1. SLC7A11的转录、表观遗传和翻译后调控机制。
已知SLC7A11的表达可以在多种应激条件下被诱导,包括氧化应激,氨基酸饥饿,代谢应激和遗传毒性应激,可能是一种适应性反应,使细胞能够在应激条件下恢复氧化还原稳态并维持存活。激活转录因子4(ATF4)和/或核因子红系相关因子2(NRF2)是调控压力诱导的SLC7A11表达的两个主要转录因子。此外,p53可抑制SLC7A11的转录,从而调节铁死亡,在肿瘤抑制中起关键作用。而ATF3也可以与SLC7A11启动子结合并抑制其表达。
BAP1是一种核去泛素酶(DUB),甘波谊团队2019年的研究发现,癌细胞中BAP1缺乏会导致SLC7A11上调和铁死亡抵抗。值得注意的是,SLC7A11不仅可以通过mTORC1进行调控,还可以通过mTORC2进行磷酸化调控。其他研究还表明,SLC7A11的细胞表面定位也受到调节,如EGF受体(EGFR)与SLC7A11相互作用并在质膜上维持其适当的定位。最后,作者总结了关于组蛋白修饰和染色质重塑在控制SLC7A11表达和铁死亡中关键作用的最新研究。
2
SLC7A11部分通过抑制铁死亡促进肿瘤的发展
作者全面分析了SLC7A11在抑制铁死亡、促进肿瘤发展中的作用。各种研究表明肿瘤抑制因子(如p53和BAP1)的丧失,原癌基因的突变(例如KRAS)或促肿瘤功能蛋白的过表达(例如OTUB1)会增加SLC7A11(通过上调其转录水平或稳定其蛋白质)的水平,从而抑制导致铁死亡并促进肿瘤的发展(图2)。
然而,并非所有肿瘤都显示SLC7A11水平升高,有些肿瘤(例如ARID1A缺陷型肿瘤或p53功能获得性突变的肿瘤)甚至表现出SLC7A11水平降低。但是在后一种情况下,SLC7A11水平的降低显然不能解释相应癌症的肿瘤表型。相反,根据这一现象可以提出以下假设,即SLC7A11的低表达为SLC7A11低表达肿瘤创造了致命弱点并可以用作治疗的靶向,因为这些肿瘤可能更容易受到氧化应激的影响。
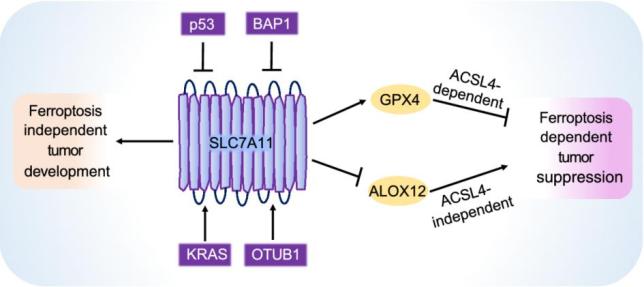
图2. SLC7A11通过铁死亡依赖性和铁死亡非依赖性机制促进肿瘤的发展。
3
SLC7A11通过铁死亡非依赖性功能促进肿瘤的发展
虽然SLC7A11在抑制铁死亡中的作用已得到很好的确立,但其他研究表明SLC7A11也可以通过其他机制促进肿瘤发展(图2)。比如,SLC7A11的失活也可以诱导细胞凋亡。目前已经确定SLC7A11可促进癌细胞的药物抗性、化学抗性和放射抗性。SLC7A11的过表达与各种治疗药物(例如顺铂、吉西他滨和MAPK途径抑制剂)的抗性相关或功能上增强了肿瘤细胞对这些药物的抗性。此外,有研究表明辐射可以诱导SLC7A11表达,SLC7A11的过表达促进肿瘤的放射抵抗,而SLC7A11的抑制则增强癌细胞或肿瘤的放射敏感性。而近期的研究证实,SLC7A11主要通过抑制铁死亡来促进放射抗性。有趣的是,最新的研究表明SLC7A11介导的谷氨酸输出可与肿瘤免疫系统相互作用并调节肿瘤免疫系统,进而影响肿瘤的生长和对癌症疗法的反应。
综上所述,SLC7A11在肿瘤生物学中铁死亡非依赖性的功能包括其对其他非铁死亡式细胞死亡、细胞增殖、细胞入侵、化学/药物/放射抗性和肿瘤免疫力的调节,以上功能可以通过SLC7A11介导的胱氨酸摄取和/或谷氨酸输出来实现。
4
SLC7A11诱导癌细胞的营养依赖
已知许多癌细胞通过上调SLC7A11水平来增强其获得细胞外胱氨酸的能力,然而,新兴证据表明,这对于SLC7A11高表达的癌细胞而言要付出高昂的代价。作者认为,这些“成本”主要来自于SLC7A11介导的胱氨酸摄取相关的两个代谢特征。首先,作为反向转运蛋白,SLC7A11介导的胱氨酸摄取与谷氨酸输出结合。另外,由于氧化性细胞外环境,细胞外胱氨酸比细胞外半胱氨酸稳定得多,导致细胞外胱氨酸的浓度比细胞外半胱氨酸的浓度高得多。为了获得半胱氨酸,癌细胞主要依靠摄取细胞外的胱氨酸(而不是细胞外的半胱氨酸),然后需要将细胞内的胱氨酸转化为半胱氨酸(图3A)。由于将胱氨酸还原为半胱氨酸会消耗NADPH,因此细胞需要首先进行氧化还原“投资”(以NADPH的形式),以获得用于维持氧化还原稳态的半胱氨酸。因此,这两个与SLC7A11介导的胱氨酸摄取相关的“成本”(即谷氨酸输出和用以还原胱氨酸的NADPH消耗)使得SLC7A11高表达的癌细胞高度依赖谷氨酰胺和葡萄糖(图3)。
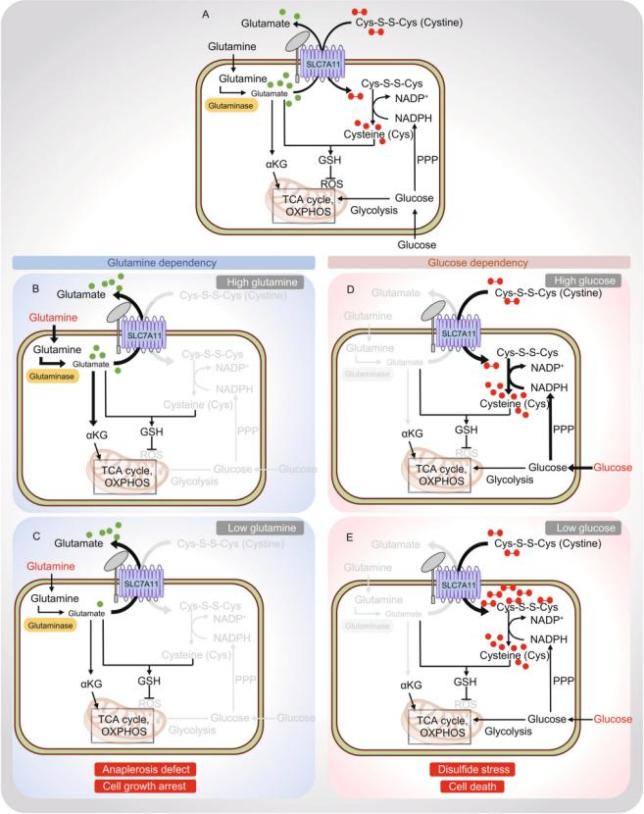
图3. SLC7A11调节癌细胞的葡萄糖和谷氨酰胺依赖性。
谷氨酸通过SLC7A11输出到细胞外,以交换细胞外胱氨酸。在SLC7A11高表达的癌细胞中,高谷氨酸输出导致细胞内谷氨酸的部分消耗,然后通过反馈抑制驱使细胞吸收更多的谷氨酰胺,并激活谷氨酰胺酶以补充谷氨酸,从而导致SLC7A11高表达的癌细胞对谷氨酰胺的依赖性(图3B-C)。因此,SLC7A11高表达的癌细胞对谷氨酰胺饥饿更加敏感。
葡萄糖是支持癌细胞存活的主要营养物质之一。许多研究都表明,SLC7A11高表达的癌细胞高度依赖葡萄糖生存,同时对葡萄糖饥饿诱导的细胞死亡更加敏感。而SLC7A11的失活则显着抑制了葡萄糖饥饿导致的细胞死亡。总之,以上研究表明SLC7A11促进癌细胞的葡萄糖依赖性。
那么SLC7A11如何促进葡萄糖依赖性?甘波谊在此处介绍了其团队的最新研究结果,并综合了近几年其他研究团队的发现,解释了SLC7A11在癌细胞的葡萄糖依赖性中的作用机制。在SLC7A11高表达的癌细胞中,大量的细胞外胱氨酸被摄入细胞。由于其低溶解度,细胞内胱氨酸的积累可能具有毒性,从而迫使细胞迅速将胱氨酸还原为可溶性更高的半胱氨酸。由于该反应需要NADPH,并且胞质NADPH主要通过葡糖糖-戊糖磷酸途径 (PPP) 提供,因此SLC7A11高表达的癌细胞表现出葡萄糖-PPP依赖性。限制SLC7A11高表达癌细胞中的葡萄糖,可导致NADPH耗竭,使得细胞内胱氨酸和其他二硫化物分子积累,从而导致二硫化物应激和细胞快速死亡(图3D-E)。
综上所述,这些研究表明,SLC7A11过表达的癌细胞通过不同潜在机制诱导谷氨酰胺和葡萄糖依赖性。值得注意的是,SLC7A11高表达的癌细胞对谷氨酰胺或葡萄糖缺乏的细胞表型似乎有些不同:谷氨酰胺缺乏(或谷氨酰胺酶抑制)主要抑制SLC7A11高表达癌细胞的细胞生长,葡萄糖饥饿导致SLC7A11高表达癌细胞的快速死亡(这与这种情况下的严重氧化还原系统崩溃相一致)。当利用N-乙酰半胱氨酸(NAC)来挽救葡萄糖不足导致的细胞内氧化还原时,NAC很大程度上挽救了SLC7A11高表达癌细胞的细胞死亡,但不能挽救这些细胞在谷氨酰胺剥夺或谷氨酰胺酶抑制后的生长缺陷。
4
针对SLC7A11的癌症治疗策略
正常细胞或组织可以通过从头获得细胞内半胱氨酸来补偿SLC7A11的损失、半胱氨酸合成或通过其他转运蛋白摄取胱氨酸(或半胱氨酸)。而癌细胞与正常组织相比,它们更依赖SLC7A11介导的胱氨酸摄取来获取半胱氨酸并维持氧化还原稳态。SLC7A11这两个特征,即其在正常生理中的非必需性和在癌症中的高表达,表明靶向SLC7A11可能会选择性杀死肿瘤细胞并损害肿瘤生长,同时保留正常细胞或组织,因此将SLC7A11提名为有希望的癌症治疗靶点。
作者将目前针对SLC7A11的癌症治疗的临床前研究进行了总结,并将其概念化为两种广泛的策略(图4)。第一个涉及直接抑制SLC7A11转运蛋白的活性,而另一个涉及针对癌症中与SLC7A11相关的代谢敏感性(葡萄糖或谷氨酰胺依赖性)。(1)使用各种抑制剂直接阻断SLC7A11胱氨酸转运蛋白的活性。这些药物抑制SLC7A11吸收胱氨酸,从而诱导脂质过氧化和铁死亡。(2)通过抑制葡萄糖摄取来靶向SLC7A11高表达癌细胞的葡萄糖依赖性。SLC7A11高表达癌细胞中可用葡萄糖的降低会诱导二硫键应激,导致细胞快速死亡。(3)针对SLC7A11高表达癌细胞中的谷氨酰胺依赖性,通过使用谷氨酰胺酶抑制剂(例如CB-839)来抑制癌细胞的生长。
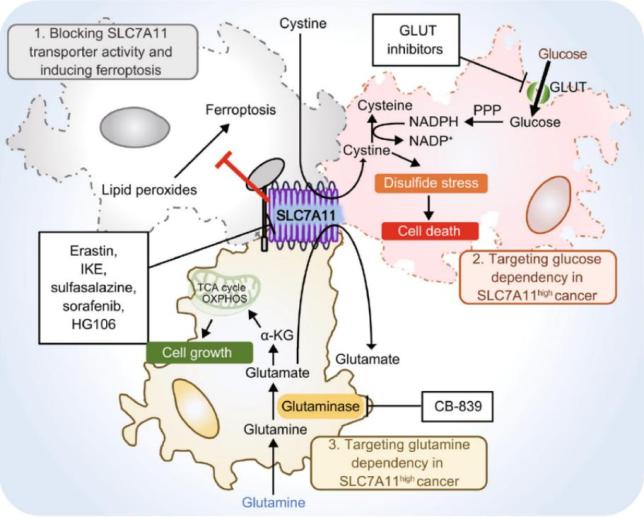
图4.靶向SLC7A11的癌症治疗策略。
结论和未来展望
癌细胞通过多种机制上调SLC7A11的表达,从而增强其抗氧化防御能力并抑制铁死亡,这对肿瘤的生长是有益的。然而,建立半胱氨酸源性抗氧化防御系统给SLC7A11高表达癌细胞带来了一个巨大的成本(包括谷氨酸输出、胱氨酸摄入和NADPH供应以将细胞中的胱氨酸还原为半胱氨酸),导致葡萄糖和谷氨酰胺依赖性。由于葡萄糖和谷氨酰胺是在细胞外环境中最丰富的营养物质中,在正常情况下,SLC7A11高表达癌细胞可以吸收足够量的葡萄糖和谷氨酰胺来满足其抗氧化防御的这些要求。基于此,作者提出在大多数情况下,SLC7A11主要发挥肿瘤促进作用,但其过度表达的确暴露了癌细胞的致命弱点(使细胞对葡萄糖或谷氨酰胺饥饿更为敏感)。因此,作者进一步认为,在葡萄糖或谷氨酰胺限制条件下,SLC7A11甚至可能发挥肿瘤抑制作用,但这一假设仍需要进一步检验。
同时,作者对癌症中SLC7A11的未来研究方向,提出以下几点建议。
1. SLC7A11的翻译后调控。我们对SLC7A11翻译后水平上调控的了解仍然处于初级阶段。此外,这种翻译后调控反过来如何调节SLC7A11介导的下游生物学效应,如铁死亡和营养依赖性,仍然是未知的。由于SLC7A11促进葡萄糖饥饿时的细胞死亡,因此测试mTORC2或AKT介导的SLC7A11抑制性磷酸化是否可抑制SLC7A11响应葡萄糖饥饿,从而在葡萄糖饥饿的条件下促进细胞存活,将是一个有趣的方向。以上这些有趣的问题都需要进一步研究。
2. SLC7A11在肿瘤微环境中的功能。考虑到SLC7A11从细胞外环境摄入胱氨酸和向外输出谷氨酸的功能,SLC7A11可能在介导肿瘤细胞与肿瘤微环境之间的相互作用中发挥作用。然而,当前大多数研究集中在肿瘤细胞中SLC7A11的细胞自主功能。作者设想,将来的研究需要同时阐明SLC7A11在肿瘤生物学中的潜在细胞非自主功能。同样,SLC7A11是否以及如何在免疫细胞或基质细胞中发挥功能,进而调节肿瘤细胞的行为,也是未来研究潜在的迷人话题。
3. 胱氨酸还原酶的确证。消耗NADPH以将胱氨酸还原为半胱氨酸的胱氨酸还原酶的身份仍然难以捉摸。因此需要进一步的遗传学研究确定真正的胱氨酸还原酶。
4. 如何推动靶向SLC7A11肿瘤治疗策略的临床应用。为了将针对SLC7A11的肿瘤治疗策略应用于临床,作者建议在未来的临床前研究中应进一步解决以下两个问题。
第一个问题是确定用于靶向治疗的特定肿瘤或基因型背景。已有研究表明具有某些致癌突变的肿瘤(例如KRAS突变肿瘤)依赖于SLC7A11介导的胱氨酸摄取,因此此类肿瘤可能对SLC7A11抑制剂特别敏感。
第二个问题是研究并确立与靶向SLC7A11的各种抑制剂的合理组合策略,以实施更有效的靶向治疗。如抑制剂和免疫治疗、放射性治疗的合理联合方案的开发。
原文链接:
https://doi.org/10.1007/s13238-020-00789-5
本文转载自公众号“BioArt”(BioGossip)
由中国生物物理学会主办的
“第十八次中国暨国际生物物理大会”
将于2020年11月15-18日
在广东省广州市召开
欢迎全国各研究机构、高等院校
和企业的科技工作者参加
中国生物物理学会官方订阅号,为BSC会员及生物物理领域专业人士服务。
投稿及授权请联系:bscoffice@bsc.org.cn。
微信号:BSC-1979
喜欢此内容的人还喜欢
原标题:《【科技前沿】Protein & Cell:甘波谊团队全面分析SLC7A11/xCT在癌症中的作用——铁死亡,营养依赖,肿瘤治疗》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2025 上海东方报业有限公司

