- +1
使用B细胞和T细胞受体序列的机器学习进行疾病诊断
设计了实验方案和数据分析框架,用于识别与感染性疾病、免疫性疾病或疫苗接种等干预措施相关的人类BCR重链和TCRβ链特征。该方法名为“免疫诊断机器学习”(Mal-ID),结合了传统免疫学分析(如检测同一疾病个体间共享序列)与人工智能(AI)蛋白质序列模型(称为蛋白质语言模型)衍生的复杂特征。尽管AI系统的决策过程可能难以解释,但该团队开发了理解模型诊断预测原理的方法。
技术实现路径如下图所示:
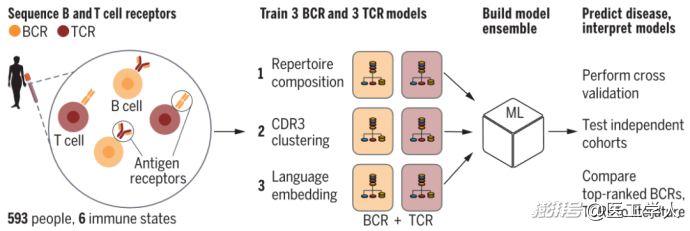
从血液样本到疾病分类的免疫受体测序流程:对593个体进行B/T细胞受体测序后,通过(1)受体群体/“库”组成分析;(2)决定抗原特异性的CDR3序列区域聚类;(3)蛋白质语言建模,最终基于BCR/TCR信息实现高精度疾病分类(交叉验证中取得优异的多分类AUROC评分)。
一、Mal-ID技术框架
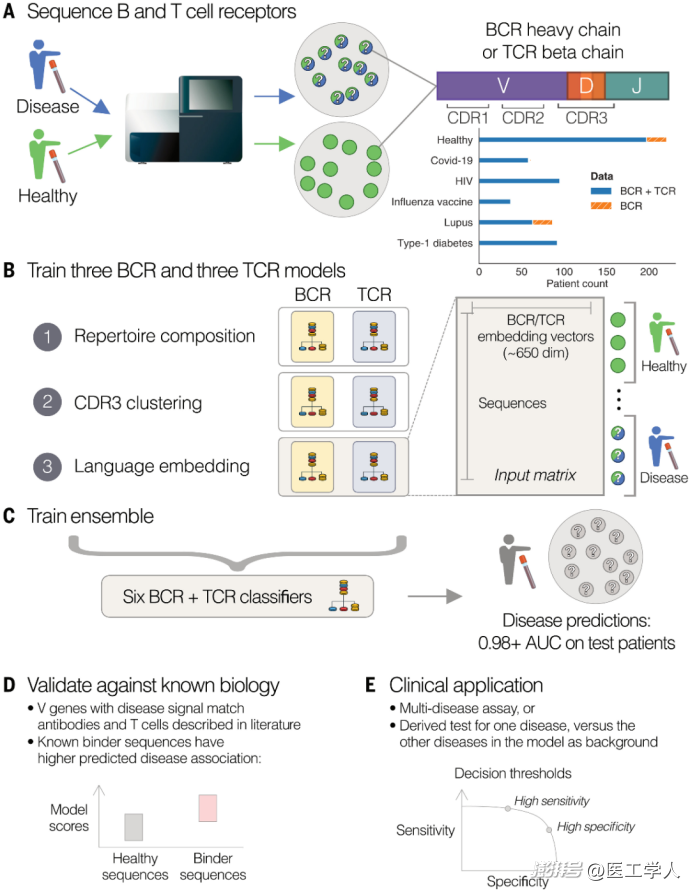
图1. Mal-ID技术框架
首先从不同疾病状态患者的血液样本中获取BCR重链和TCRβ链的基因库数据(A部分);随后采用三类特征模型进行分析——包括基因片段使用频率与突变率(模型1)、CDR3序列聚类(模型2)和基于蛋白质语言模型的CDR3结构预测(模型3)(B部分);通过集成6个基础模型(3个BCR+3个TCR)构建逻辑回归分类器,实现对保留测试集的疾病概率预测(C部分);该框架支持验证V基因的疾病特异性信号(D部分),并可根据临床需求灵活调整为多病筛查或单病诊断模式(E部分)。整个流程通过整合免疫受体库的多维度特征,实现了高精度(AUROC 0.986)的免疫状态分类。
二、联合分析BCR和TCR数据
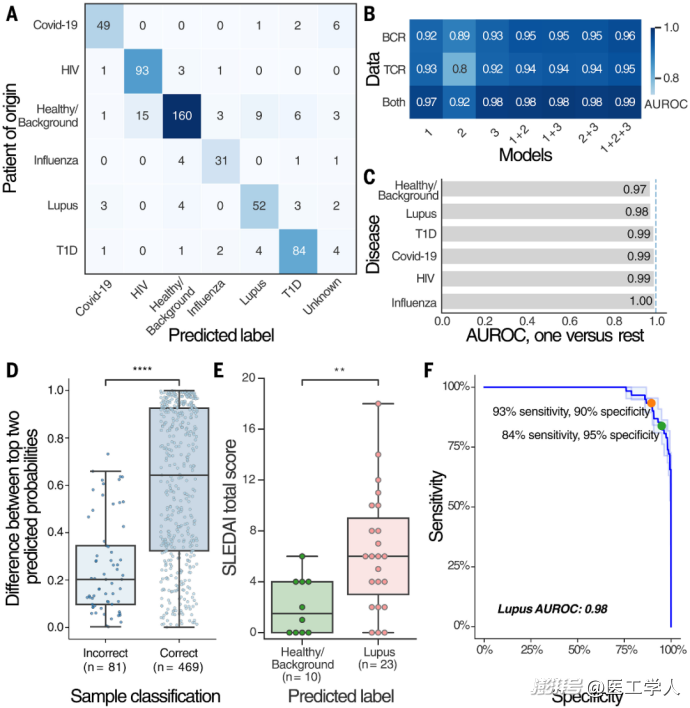
图2. Mal-ID通过IgH和TRB序列进行疾病分类
通过整合B细胞受体(IgH)和T细胞受体(TRB)序列数据进行疾病分类的综合性能:集成模型在550个独立测试样本中实现高精度分类(A),多模型比较显示结合BCR和TCR数据的集成方法(AUROC0.98)显著优于单一模型或单数据类型(B);各疾病类别分类效能均衡(C),且模型对正确预测结果具有更高置信度(D)。特别地,成人狼疮患者的误分类与较低临床活动指数(SLEDAI)相关(E),表明模型可能捕捉到治疗缓解期的免疫特征变化。此外,从多疾病分类器衍生的狼疮专用诊断模型可灵活调整阈值实现93%灵敏度/90%特异性的平衡性能(F)。该结果验证了联合分析BCR和TCR数据对提升免疫疾病诊断可靠性的关键作用。
三、语言模型重现免疫学知识
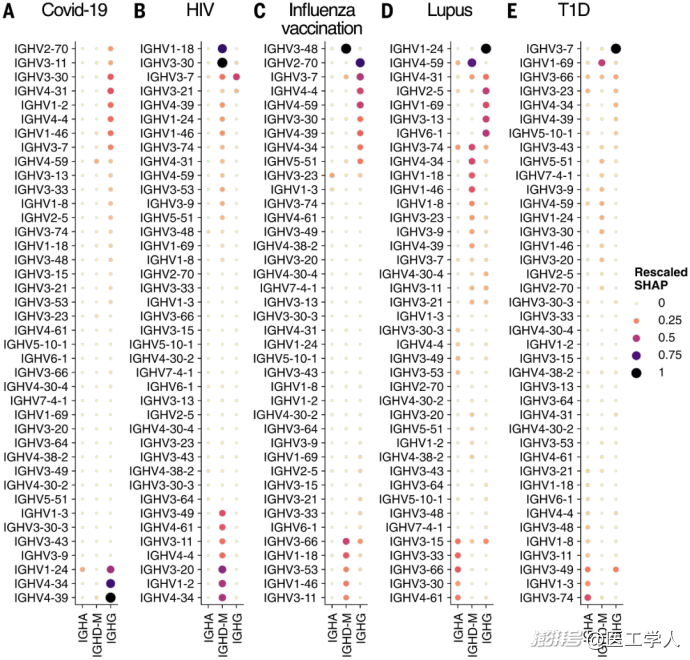
图3. 模型3通过蛋白质语言嵌入识别的疾病相关IGHV基因与同种型
模型3通过蛋白质语言嵌入技术识别的疾病特异性IGHV基因和同种型特征:基于SHAP值分析显示,COVID-19预测主要依赖IGHV1-24/IGHV2-70基因与IgG同种型(A),HIV与突变型IgM/D和IGHV1-2/IGHV4-34基因显著相关(B),流感疫苗接种响应集中于IGHV3-23基因及IgG/突变型IgM/D(C),而狼疮和1型糖尿病(T1D)则分别与IGHV4-34/IGHV4-59基因及IgA(D)、多种同种型特征(E)具有强关联。这些发现不仅与已知的病原体特异性抗体反应(如SARS-CoV-2的IgG优势)和自身免疫病机制(如狼疮的IgA自身抗体)相吻合,更通过量化不同免疫球蛋白类型的贡献,揭示了疾病特异性的B细胞应答模式。
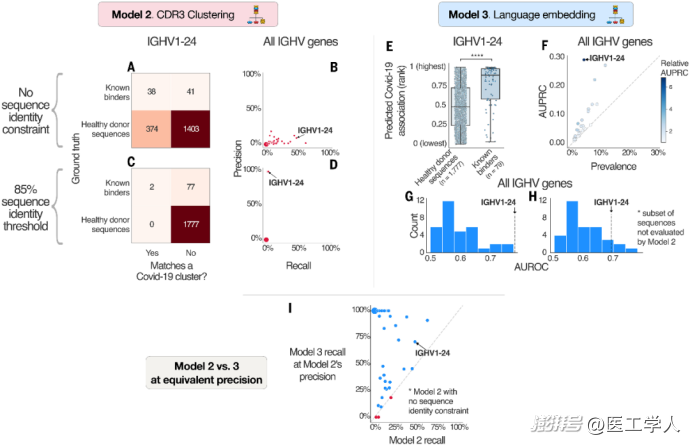
图4. 模型2与模型3从COVID-19患者数据中
学习SARS-CoV-2抗原特异性序列模式
Mal-ID的模型2(CDR3聚类)和模型3(蛋白质语言模型)能够从COVID-19患者数据中自主识别SARS-CoV-2抗原特异性抗体序列特征:模型2通过保守的公共克隆识别(IGHV/IGHJ基因和CDR3长度匹配)可精确检测部分已知结合序列(A-D,如IGHV1-24基因中达100%精确度但召回率低),而模型3则展现出更全面的识别能力——在未接触训练数据的情况下,对CoV-AbDab数据库中已验证的SARS-CoV-2结合序列的预测概率显著高于健康供体序列(E,置换检验P=0),跨IGHV基因的AUROC最高达0.78(G),且对模型2未覆盖的序列仍保持有效区分(H,AUROC≦0.75)。两种模型互补性强:模型3在相同精确度下比模型2召回率更高(I),但需权衡更多假阳性,这一发现揭示了机器学习可从复杂免疫受体库中提取抗原特异性信号,为病原体特异性抗体发现提供了新范式。
最后,作者强调使用来自大量患者的经验数据,这些患者持续收集了IgH和TRB免疫受体测序数据,这些数据伴随着对批次效应和混杂因素的潜在担忧,并试图解决这些问题。对所有样本使用了标准化的受体测序方案和生物信息学分析,并确定基于人口统计学协变量的模型无法像IgH和TRB特征那样准确地对患者免疫状态进行分类。从主要分析中保留了患者队列,并确认他们在验证步骤中被正确分类。来自其他实验室的完全独立队列的性能进一步表明,Mal-ID 泛化于独立数据,不适合潜在的、未知的隐藏变量。Mal-ID 框架似乎捕获了免疫反应的基本原理并推广到单独的临床队列。区分Covid-19、HIV感染、狼疮、T1D和健康的任务被用来证明该方法的潜力。需要额外的测试,以便在临床研究中确定对具有不同和可变患病率的特定疾病的敏感性和特异性的适当临界值,并进一步评估最佳样本量和测序深度。该方法的任何结果都需要根据患者的其他临床评估和实验室测试进行解释。其他需要解决的重要主题是同一患者可能存在多种疾病或合并症,针对特定疾病的不同严重程度或亚型的模型开发,使用其他种类含有淋巴细胞的标本(如组织活检)的价值,以及确定先前模型中未包含的疾病证据的可能性, 例如在未来大流行中可能发生的事件。
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2026 上海东方报业有限公司

