- +1
识病寻源|一家四代十名男性总共12颗牙,此病至今无准确发病率
·从达尔文对外胚层发育不全综合征(ED)第一次描述至今,人们认识这一疾病已超一个半世纪。从临床特征鉴定、流行病学描述,到分子机制研究、动物实验,直到临床研究,我们见证了一个罕见病的认识-研究-治疗进程。在这一进程中,需要多方的积极参与:相互扶持的患者、无私负责的医生、深入探索的科学家、给予支持的基金会、有社会责任感的制药厂商,最后,可能还需要一个文明社会的高度关注与包容。
2022年,EDA羊膜内注射的多中心临床试验正式启动,代号EDELIFE。图片来源:https://edelifeclinicaltrial.com
1831年12月,一艘单桅帆船在英国普利茅斯港整装待发。按计划,这艘船将赴南美洲调查,之后环球航行返回英国。一位22岁的自然史爱好者抓住了这个机会,他愿意忍受船上的艰苦环境,由海军提供食宿,借机拜访这些遥远的国度。他登上船只时,并没有意识到这次旅程将成为科学史上最伟大的航程之一,而他自己也将因此名垂青史。
年轻的查尔斯·达尔文(Charles Darwin,1809-1882)就这样登上了港口的“小猎犬号”,远赴南美洲,详尽地采集动植物样本并编目。1835年,“小猎犬号”离开南美洲,耗费超过一年的时间绕行太平洋及非洲大陆,在塔希提岛、新西兰、澳洲、 科科斯群岛、开普敦、圣赫勒拿岛等地停靠,最终在1836年10月回到英国法尔茅斯。
达尔文在环球旅行中搜集的大量标本与笔记,为他思想体系的成熟奠定了条件。他构思了一项宏伟的计划,将他的理论作为多卷本出版,第一本便是著名的《物种起源》,紧随其后开始撰写的第二本专著《动物和植物在家养下的变异》知名度略低。
《动物和植物在家养下的变异》的第一卷涉及家养品种之间的差异,强调物种的可塑性。第二卷讨论了他对遗传的理解。有意思的是,在这本书中,达尔文主要基于他渊博的知识储备讨论物种的变异,但他很可能无意间第一次用文字记录了一类遗传病的临床特征。
达尔文在这本书中提到:一个印度家系四代之中,十名男性加起来仅有4颗很小的门牙和8颗后磨牙[1]。根据这样的描述,学者们推测,达尔文很可能在谈论一类遗传病:外胚层发育不全综合征(ectodermal dysplasia, ED)。

查尔斯·达尔文《动物和植物在家养下的变异》。图片来源:《The variation of animals and plants under domestication》
外胚层发育不全综合征(也称外胚叶发育不全)不是一项疾病,而是由200多种疾病组成的一系列疾病的统称。人类在胚胎发育早期,会形成内、中、外三个胚层,它们各自发育为特定组织和器官。其中外胚层主要发育为表皮及其附属结构,如汗腺、毛发、牙齿、指甲等。ED的主要临床特征就是这些外胚层衍生物发育不良或缺失。
在1929年,该病第一次见诸学术期刊,报道了一名伦敦患儿具有头发和牙齿发育不良的体征[2]。从1970年代开始,学术界开展了系统性地研究,首先定义了ED的经典体征,即影响两种或两种以上的外胚层衍生物(如头发、牙齿、指甲、某些腺体)等,并区分出上百种临床表现形式[3]。患者的体格外观特征有:头发稀疏、色素减退、指甲发育不良、低位耳、嘴唇外翻、汗腺缺失、皮脂腺缺失、皮肤角化、牙齿缺失等等。其中很多特征在幼儿阶段难以辨别,而患者常因牙齿缺失前往口腔科就诊。所以,ED易于在口腔科门诊中被发现,是受到口腔科医师关注的一类遗传疾病。
由于ED的表现形式多样,遗传因素复杂,迄今都没有准确的发病率数据。但据估计,每100,000名婴儿中就有7例受ED的影响。2013年丹麦的一项流行病学研究报道,ED中较为常见的一类X连锁无汗型外胚层发育不全(X-linked hypohidrotic ectodermal dysplasia, XLHED)发生率为1.6 : 100,000[4]。
制定临床诊断标准后,科研人员逐步深入探索该病的分子遗传致病机制。1996年,芬兰科学家通过对多个家系的分析,确定了第一个ED相关的遗传突变,成功克隆了X连锁无汗型外胚层发育不全的致病基因。该基因位于X染色体上,编码一种跨膜蛋白EDA[5],并在人皮肤各类附属器官中大量表达。3年后,美国科学家在2号常染色体上找到另一个致病基因EDAR,后来发现是EDA的受体[6]。这提示EDA相关的信号通路在皮肤衍生物发育中的重要作用,这条通路的破坏是导致ED的原因之一。在小鼠中,如果突变这条信号通路,新生的小鼠显示出毛发缺失的表型。
在随后的研究中,EDA-EDAR这一配体-受体信号通路被阐述的更加清晰,是目前研究最深入的ED致病机理之一。除此之外,还有许多信号通路参与了ED的发生。比如NF-κB信号通路,细胞粘附相关分子如角蛋白、Nectin1等,以及影响胚胎发育的Wnt信号通路等。各类信号之间还可能存在串扰。这些复杂的发病机制造就了ED多种不同的临床表征,已经得到了全面的总结和讨论[7]。然而,仅仅使用临床表征来诊断ED并不准确。当前对ED的诊断逐步过渡到分子诊断,基于人类遗传数据库和疾病数据库,配合分子检测及遗传咨询师的帮助,可较为准确地诊断ED。当前可进行分子检测的ED类型已逾60余种[7]。
作为一类遗传病,目前尚无针对ED的有效疗法。临床上主要是缓解症状。ED引起的皮肤干燥等问题主要通过保湿霜等皮肤护理产品处理。对于汗腺缺乏,主要是减少运动并通过物理方法降温。对于牙齿缺失,可考虑制作活动义齿或进行种植修复。
需要注意的是ED患者的心理问题,由于面部结构的异常,患者可能受到歧视并患有心理障碍。这需要心理治疗和社会的扶助,重视患者的情感诉求和社交需求。当前的医学可能在一定程度上能协助修复患者的生理功能,但如何确保残障人士享有同等的权利和自由,践行联合国残疾人权利国际公约,是全社会应当考虑的关键议题。
显然,科学家并不满足当前“治标”式的疗法,仍在不断探索ED的治疗方案。根据ED的发病机制理,若要开展基因治疗,可能需要在出生前进行干预,但风险较高。目前,对于配体表达不足导致的ED,在发育过程中给予EDA重组蛋白已经成为很有前景的新疗法。这种疗法并不改变患儿的DNA,而是在发育过程中用药物校正。在ED动物模型中,给新生的动物注射重组EDA蛋白。未经治疗的动物只有一些不完整的畸形牙齿,而治疗后牙齿发育良好,流泪和出汗的能力也得到了提高[8]。
那么如果在动物发育的更早期给予这个蛋白,会不会具有更好的效果?在小鼠模型中,科学家改造了EDA蛋白的结构,令其能通过胎盘屏障。在胚胎发育过程中接受这种治疗的小鼠出生后,毛发、牙齿等的表型与健康小鼠非常相似[9]。看来使用EDA蛋白治疗这种配体表达不足导致的发育性疾病是可行的。

ED小鼠的尾巴。左:未经治疗的小鼠,右:在胚胎发育过程中接受治疗的小鼠。图片来源:《Nat Med》
2012年,FDA批准使用重组融合蛋白Fc-EDA进行XLHED的临床研究。临床试验结果表明,Fc-EDA具有良好的安全性,但对产后才开始治疗的新生儿患者无效。在2018年,《新英格兰医学》发表了一项报道,德国儿科专家Holm Schneider等人测试了更早期的给药方法。他们对1名孕妇进行了羊膜内给药,让药物直接作用于胚胎发育过程中的胎儿。结果发现2名出生的婴儿在14-22个月中未出现相关病征,而其未用药的兄长则具有显著的疾病表现[10]。直到今天,这些接受治疗的男孩均不需要任何ED相关的住院治疗。这项工作开启了子宫内给药治疗ED的新篇章,为该疗法走向临床提供了重要的支持。
在专注于罕见病治疗的基金会、患者合作组织、以及具有社会责任感的制药公司的推动下,2022年,EDA羊膜内注射的多中心临床试验正式启动,代号EDELIFE,在德国、法国、意大利、西班牙、美国、英国开放,目前正在招募临床受试者。用于注射的药物已经获得FDA以及欧盟药品管理局的突破性疗法认定和孤儿药认定。是目前从产前校正特定类型ED的非常有希望的疗法。该项目依然由Holm Schneider等专家领衔。值得注意的是,Schneider作为XLHED产前治疗专利的发明人,主动放弃了这项发明的任何个人经济收益[11]。
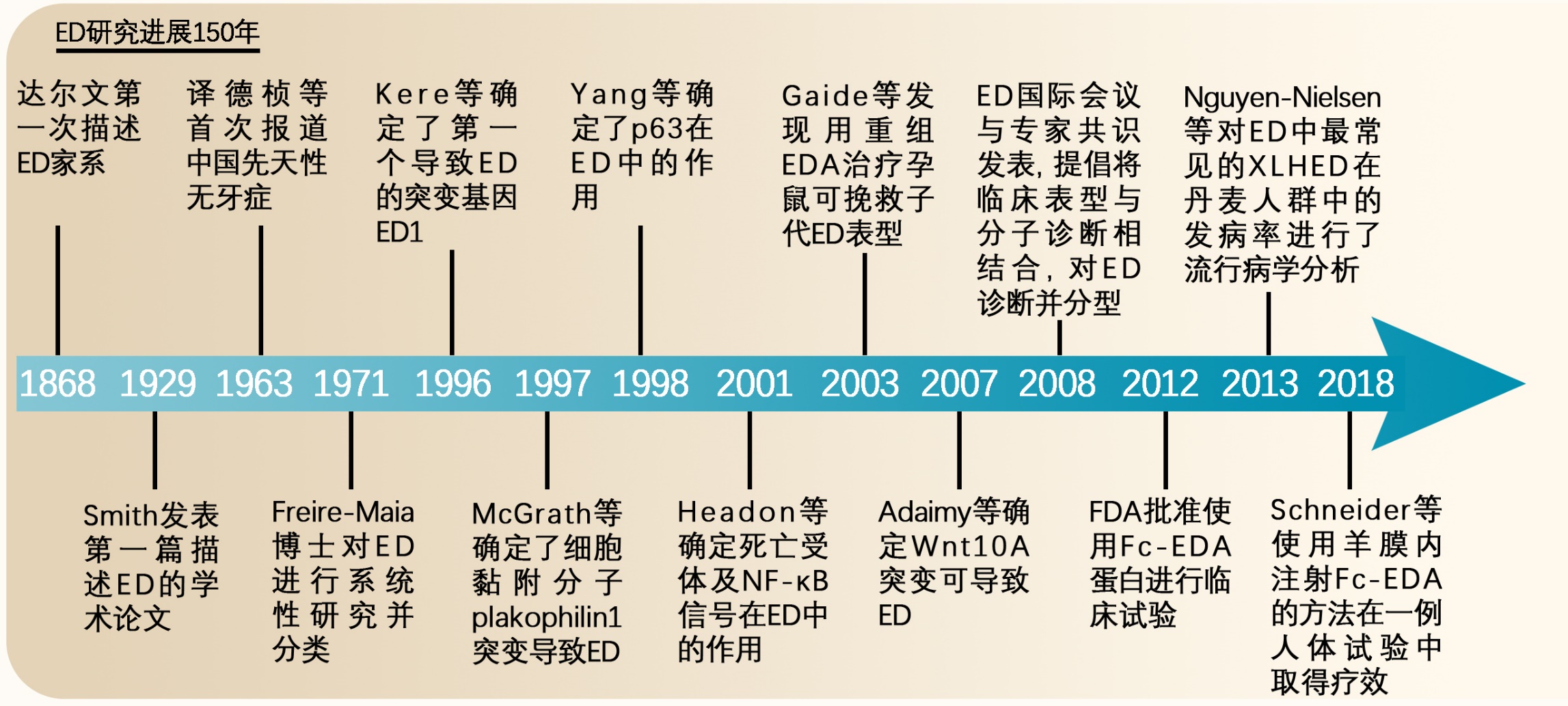
ED研究进展150年。图片来源:《中国优生与遗传》
从达尔文对ED第一次描述至今,人们认识这一疾病已超一个半世纪。从临床特征鉴定、流行病学描述,到分子机制研究、动物实验,直到临床研究,我们见证了一个罕见病的认识-研究-治疗进程。在这一进程中,需要多方的积极参与:相互扶持的患者、无私负责的医生、深入探索的科学家、给予支持的基金会、有社会责任感的制药厂商,最后,可能还需要一个文明社会的高度关注与包容。
(作者杨云龙,系复旦大学基础医学院细胞与遗传医学系研究员、副主任。叶颖,系同济大学附属口腔医院口腔种植科副主任医师、副教授。 疾病不断地改变着每个人的人生轨迹。但除了医生与医学研究者,人们很少有机会了解各式各样的疾病。“识病寻源”专栏将以一文一病的形式,介绍对疾病的认识进程,疾病的病因及其治疗。跟随医学科学的进步,理解现代医学。)
参考文献:
1. Darwin, C., The variation of animals and plants under domestication. Authorized ed. 1868, New York,: O. Judd & company.
2. Smith, J., Hereditary Ectodermal Dysplasia. Arch Dis Child, 1929. 4(22): p. 215-26.
3. Pinheiro, M. and N. Freire-Maia, Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet, 1994. 53(2): p. 153-62.
4. Nguyen-Nielsen, M., et al., The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995-2010. Eur J Med Genet, 2013. 56(5): p. 236-42.
5. Kere, J., et al., X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet, 1996. 13(4): p. 409-16.
6. Monreal, A.W., et al., Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet, 1999. 22(4): p. 366-9.
7. 叶颖, et al., 外胚叶发育不全的研究进展. 中国优生与遗传杂志, 2020. 28(03): p. 276-280.
8. Casal, M.L., et al., Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am J Hum Genet, 2007. 81(5): p. 1050-6.
9. Gaide, O. and P. Schneider, Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat Med, 2003. 9(5): p. 614-8.
10. Schneider, H., et al., Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. N Engl J Med, 2018. 378(17): p. 1604-1610.
11. Schneider, H., et al., Protocol for the Phase 2 EDELIFE Trial Investigating the Efficacy and Safety of Intra-Amniotic ER004 Administration to Male Subjects with X-Linked Hypohidrotic Ectodermal Dysplasia. Genes (Basel), 2023. 14(1).



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2026 上海东方报业有限公司

