- +1
识病寻源|从基因编辑技术CRISPR治疗失败看杜氏肌营养不良症
【编者按】2022年10月,全球唯一一名参与CRISPR基因编辑疗法的杜氏肌营养不良症(DMD)志愿者,在治疗过程中死亡。复旦大学基础医学院细胞与遗传医学系研究员杨云龙表示:CRISPR相关技术作为一种高效且广泛应用的基因编辑方法,是当前研究的热点。将其应用于临床,由于涉及到伦理和监管问题,也常常成为争议的焦点。他希望更关注DMD这一遗传性罕见病本身,DMD目前还没有治愈方法。“识病寻源”是杨云龙在澎湃科技开设的独家专栏。
2022年11月10日,《科学》杂志(Science)的新闻栏刊登了一条信息[1]:一名27岁杜氏肌营养不良症患者在一项新型基因编辑试验中死亡。虽然信息披露尚不充分,但许多科学家认为,这名患者确实通过病毒输注,接受了基于CRISPR的治疗。
根据ClinicalTrials.gov上的注册信息(NCT05514249),该临床试验仅有一名参与者。受试者2022年8月底在美国马萨诸塞大学医学院接受了静脉输注,约6周后去世。CRISPR相关技术作为一种高效且广泛应用的基因编辑方法,是当前研究的热点。将其应用于临床,由于涉及到伦理和监管问题,也常常成为争议的焦点。由于基因编辑的话题性,大量的新闻网站报道了该事件。但新闻报道中并未对受试者患有的杜氏肌营养不良症作详尽解释。例如,在美国广播公司(American Broadcasting Company, ABC)的报道中,仅有一句话描述该病:“这种罕见的遗传性肌肉萎缩疾病是由产生一种叫做肌营养不良蛋白的基因突变引起”。
杜氏肌营养不良症(Duchenne Muscular Dystrophy, DMD)得名于纪尧姆·本杰明·阿曼德·杜兴(Guillaume Benjamin Amand Duchenne, 1806-1875)。他是200年前的法国神经生物学家,进行了肌肉电刺激的开创性研究。杜兴不仅尝试用电治疗肌肉问题,还将电作为研究解剖结构和生理学的研究手段。他发明了“杜兴机”—一种用电刺激肌肉的便携装置,极大地丰富了电生理学研究。使用该装置,他发现假笑不涉及眼睛周围的肌肉,而发自内心的微笑涉及眼周和嘴部肌肉。如今,这种需要调动眼轮匝肌的真诚笑容被称为“杜兴笑”。实验过程中拍摄的电刺激面部肌肉的照片令人印象深刻,广为传播。部分原始照片现已被收入美国纽约的现代艺术博物馆(Museum of Modern Art, MoMA)
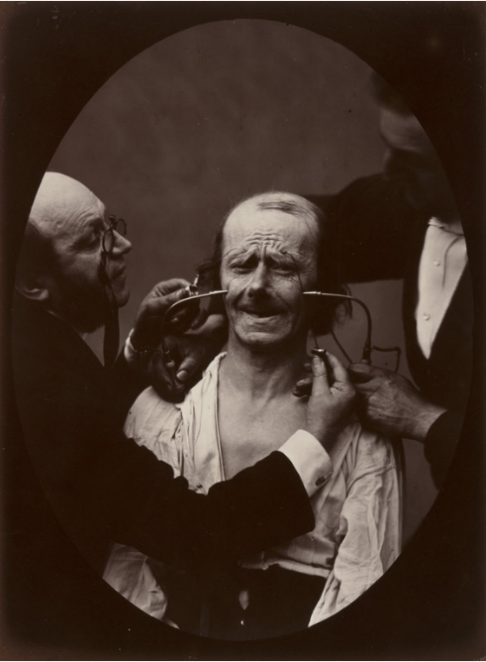
杜兴用电刺激受试者面部肌肉的照片,1856。来源:MoMA馆藏。
1842年起,杜兴在巴黎开展肌营养不良和其他神经肌肉疾病的研究,尝试用电刺激触发特定肌肉运动,并详细描述了几类神经肌肉疾病。在杜兴之前,DMD的病例已经被零星地描述过,如1830年,英国著名医生查尔斯·贝尔(Charles Bell, 1774-1842)描述了一名肌肉进行性麻痹导致下肢瘫痪的18岁男子,从10岁起大腿无力,不能站立[2]。1847年理查德·帕特里奇(Richard Partridge,1805–1873)第一次对DMD患者进行病理尸检,发现“肌肉出现脂肪变性,小腿变性程度高于上肢,神经和肌腱都没有发生变化”[3]。1851年,爱德华·梅里恩(Edward Meryon,1807–1880)首次对DMD进行了明确的临床和病理描述,他向伦敦皇家医学和外科学会提交了论文,描述患者尸检结果:“在显微镜下检查肌肉组织时,发现条纹状纤维被完全破坏,肌节成分弥散,在许多地方转化为脂滴和颗粒物,肌纤维膜被破坏”[4]。梅里恩最初认为DMD患者是由于脊髓问题导致,但经过仔细检查,他发现患者神经组织是完整的,观察到的唯一结构变化是肌纤维改变,脂肪填充了肌肉纤维鞘。从1851年到1870年,梅里恩一直对这一疾病开展研究,可能是全面总结DMD的第一人。杜兴则首次拍摄了DMD患者的照片,1862年发表在照片集《病理摄影集》(Album de photographes pathologiques)中[5],这也是首次出版的临床影像资料的书籍。更进一步,杜兴发明了一种肌肉活检针,于1864年应用,对一名DMD患者肌肉进行了活检观察。杜兴高质量地确定了DMD的关键病理结果,并在1868年总结了该病的特征。随后,神经科学家威廉·理查德·高尔斯爵士(Sir William Richard Gowers,1845–1915)在观察了多个家系后得出结论,DMD是一种具有明显男性偏好的早发性遗传疾病,其病因来自卵子。大多数病例6岁前发病,由于腿部肌肉乏力,具有特征性的站起姿势。高尔斯尝试过多种疗法,包括杜兴的电刺激疗法,未观察到疗效[6]。在确定DMD的疾病特征、病理改变的过程中,杜兴、高尔斯等人做出了大量的贡献。2003年,《神经病学年鉴》(Annals of Neurology)期刊主编Kenneth Tyler教授对DMD的早期探索历史进行了总结,并在美国神经病学年会上报告[7]。

杜兴为他第一位DMD患者拍摄的影像资料。来源:[4]
进入20世纪,DMD研究一度陷入沉寂。直到进入分子时代,大量的遗传学研究和分子生物学研究聚焦于该疾病,极大地丰富了我们对DMD的认知。通过流行病学调查,我们知道杜氏肌营养不良(DMD)是一种X连锁隐性遗传病,发病率在男性婴儿中约十万分之10,属于发病率较高的遗传病。另一类称为贝氏肌营养不良(Becker muscular dystrophy, BMD)与其发病机理类似,发病率为约十万分之8。它们在女性中极为罕见。
究其病因,DMD和BMD是由肌细胞中一个重要蛋白Dystrophin遗传改变,导致结构及功能缺失所致。编码蛋白的基因体量巨大,是人类基因组中最大的基因之一。具有79个外显子,翻译出的蛋白质量有427kDa。Dystrophin通过两端的结构域将细胞骨架蛋白F-actin与细胞外基质连接起来。此外,Dystrophin还结合肌膜、细胞骨架、通道蛋白以及信号蛋白,形成复杂的复合物,并介导一系列功能。目前对这些功能的理解还不充分。在DMD患者中,Dystrophin被截断,导致细胞骨架无法连接到细胞外基质。在BMD中,Dystrophin的关键结构域尚存,但结构不完整,所以病情较轻。遗传突变是导致Dystrophin丢失或损伤的直接原因。迄今,已在DMD和BMD患者中发现了数千种不同类型的突变。大多数的突变发生在热点区域,即3号-9号外显子以及45号-55号外显子区域。
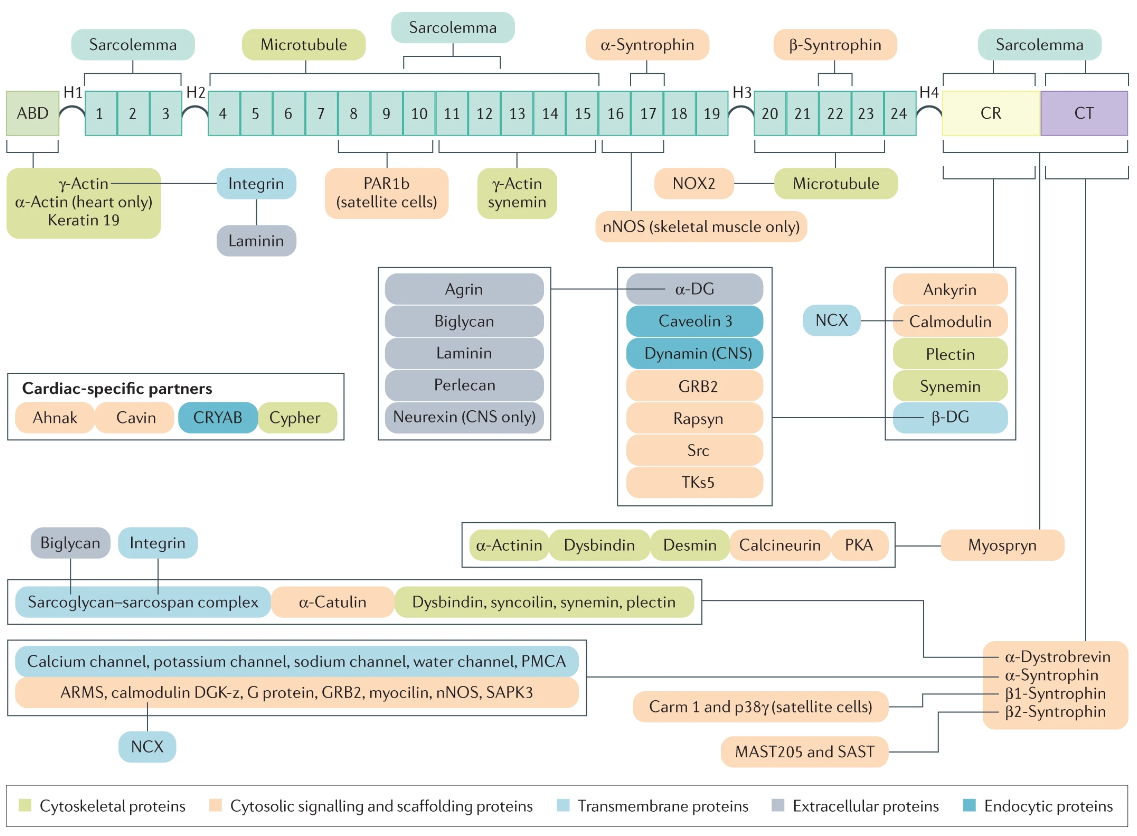
Dystrophin可以和多种蛋白结合,其缺失广泛影响肌细胞功能。来源:[8]
Dystrophin发生问题后,以其为中心的复合物会解体,肌细胞结构完整性、肌肉收缩能力、肌细胞的信号传导都会受到影响。体现在肌细胞上,就是肌膜易受收缩损伤、细胞被自由基损伤、钙离子超载、再生受阻等。这些受损的肌细胞会被炎症细胞识别并清除。在疾病早期,受损肌肉尚可通过再生修复。但在晚期,由于再生能力降低和纤维化信号上调,肌细胞被脂肪组织和纤维化组织取代,进而影响患者运动能力。
DMD目前没有治愈方法,但合理治疗可缓解疾病进展,减轻患者负担。在疾病诊断方面,若发现男孩有症状,要对患者家庭开展基因咨询,协助诊断。这有助于分型并指导未来治疗。在治疗方面,目前的标准方案是:DMD患儿在其运动发育停止时(一般在4-5岁)使用糖皮质激素,并在整个生命周期内持续治疗,这主要是由于糖皮质激素可促进肌细胞增殖同时抑制炎症。该疗法可延缓病情,增进患者生存。同时,对症的多学科治疗和高质量的护理也可改善生活质量和寿命。这包括医师、护士、康复师、营养师、心理医生等的共同合作。对于DMD的诊疗方案,2018年发表于Lancet Neurology的指南做了最为完善的总结 [9-11]。
由于DMD的不可治愈,在科研领域,科学家仍在不断寻找肌营养不良的疗法,确认Dystrophin缺失是病因后,一个朴素的“治本”的想法就是能否令肌细胞重新表达这一蛋白。由于编码蛋白的基因体量过大,直接导入全长基因并不现实。于是科学家们开发了一系列新疗法,主要包括基因疗法和细胞疗法。具体可分为以下几类:
终止密码子通读:有5%-7%的DMD病例,是由于基因突变形成了终止密码子,也就是无义突变。该突变导致Dystrophin合成提前终止。若能强制要求蛋白继续合成,就可能得到一个全长的蛋白,从而治疗疾病。一种小分子药物Ataluren可以诱导核糖体在遇到无义突变时继续向下翻译。在临床试验中,该药物未能达到主要终点,但显示了一定的治疗趋势,且安全性良好。因此得到了欧洲药监局的条件性使用批准。
外显子跳跃:蛋白表达需要先转录为mRNA,通过剪接将多个外显子拼在一起。某些特定的突变可导致蛋白合成提前终止。若能跳过该突变所在的外显子区域,产生一个不是全长的蛋白。虽然仍不及健康蛋白,但可显著减轻病情。这一方法利用寡核苷酸片段改变剪接过程,体积小,易于递送,安全性好。该方法针对不同突变要设计不同的寡核苷酸药物,目前的药物针对突变最多的群体,如外显子44、45、51、53等,需要重复治疗。其中部分药物已获美国药监局的条件性使用批准。
表达微型蛋白:使用腺相关病毒(Adeno-associated virus, AAV)可递送cDNA,令感染的组织表达特定蛋白。该方法也是其他基因疗法中常见方案。但在DMD中,由于AAV容量有限,无法运载编码Dystrophin的载体。于是科学家提出了仅将最关键结构域重组为微型蛋白,再用AAV导入的方法。临床试验结果证实,微型蛋白在受试者肌细胞中成功表达,但目前还不知道对疾病的改善情况。需要注意的是,AAV可能导致患者出现严重的免疫反应。即使对患者进行了预筛选和免疫抑制,仍有患者出现了严重不良事件。 2021年12月,辉瑞公司在一例受试者报告死亡后,暂停临床试验,目前正在恢复中。
基因组编辑技术:如果能够对基因组直接进行靶向修饰,纠正突变,或者将导致蛋白丢失的突变删除或替换,是非常有吸引力的方法。通过设计引导RNA,引入CRISPR系统,可以做到在基因组特定位点删除外显子、取消剪接位点等。该方法在动物实验中已获成功。但由于基因编辑的脱靶风险和对其中的蛋白或病毒载体产生免疫反应的风险,基因组编辑尚未进入临床。开篇提及的新闻报道中,死亡的患者可能参加了这一疗法的临床研究。
干细胞移植:将带有正常基因的干细胞移植,修复肌肉。虽然该方法有吸引力,但肌肉组织的体量对该方法提出了挑战。临床试验也发现,仅在局部注射的位置旁有一些恢复。该方法目前未获批准。
类似蛋白替代:Dystrophin有一种类似蛋白称为Utrophin,若上调其表达可产生一定的代偿效应。有临床试验研究了能够上调Utrophin的药物,但未观察到治疗效果。
除了这些“治本”的疗法,还有许多药理学药物在临床试验中,或可通过减少纤维化、减轻炎症,扩张血管、保护线粒体功能、靶向肌肉调节蛋白等不同机制,可能缓解病情[12]。
从患者的角度看来,多种疗法带来了治愈疾病的希望,但治疗方案与患者的适配性仍有待考量,且方法之间可能互斥。综合比较,表达微型蛋白是一种普适的方法,但其仍存在安全隐患。其不良反应可能主要由AAV介导,涉及制造过程中的诸多因素,如剂量、AAV类型、转入的载体、患者是否已有抗体等。开篇提到的新闻报道中,患者应该是选择了基于CRISPR的基因组编辑技术。报道体现了公众对以CRISPR为代表的基因疗法的担忧。在该试验更多信息披露之前,更重要的可能是提高公众对疾病和各类疗法的深入理解。更进一步,患者和患者家属通过理解相关研究,决定治疗方案,决定是否参与前沿疗法的临床研究,需要勇气、知识、智慧和积极的人生观。而对于患有DMD这样的罕见病患者,在某种程度上,积极的人生观要比新颖的疗法更为重要。
(作者杨云龙,系复旦大学基础医学院细胞与遗传医学系研究员、副主任。疾病不断地改变着每个人的人生轨迹。但除了医生与医学研究者,人们很少有机会了解各式各样的疾病。“识病寻源”专栏将以一文一病的形式,介绍对疾病的认识进程,疾病的病因及其治疗。跟随医学科学的进步,理解现代医学。)
参考文献
1. https://www.science.org/content/article/news-glance-new-antibiotic-covid-19-antarctica-and-venus-mission-deferred. doi: 10.1126/science.adf7363
2. Bell, C., The nervous system of the human body : embracing the papers delivered to the Royal Society on the subject of the nerves. 1830, London: Longman, Rees, Orme, Brown, and Green. xxiii, 238, clxxvi p., 9 leaves of plates.
3. Patridge R. Fatty degeneration of muscle. Med Times Gaz 1847; 5: 944.)
4. Meryon, E., On Granular and Fatty Degeneration of the Voluntary Muscles. Med Chir Trans, 1852. 35: p. 73-84 1.
5. Duchenne GBA. Album de photographes pathologiques. Paris: Bailliere; 1862.
6. Gowers WR. Clinical lecture on pseudo-hypertrophic muscular paralysis. Lancet 1879; 2: 1–2, 37–39, 73–75, 113–116.
7. Tyler, K.L., Origins and early descriptions of "Duchenne muscular dystrophy". Muscle Nerve, 2003. 28(4): p. 402-22.
8. Duan, D., et al., Duchenne muscular dystrophy. Nat Rev Dis Primers, 2021. 7(1): p. 13.
9. Birnkrant, D.J., et al., Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol, 2018. 17(3): p. 251-267.
10. Birnkrant, D.J., et al., Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol, 2018. 17(4): p. 347-361.
11. Birnkrant, D.J., et al., Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol, 2018. 17(5): p. 445-455.
12. Markati, T., et al., Emerging therapies for Duchenne muscular dystrophy. Lancet Neurol, 2022. 21(9): p. 814-829.



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2025 上海东方报业有限公司

