- +1
【重磅综述】心脏衰老的分子机制及干预靶点
以下文章来源于老顽童说 ,作者老顽童说
老顽童说.
公众号致力于传播衰老相关的前沿科研进展和趣味科普,帮助大家更深入地了解衰老背后的科学故事~

关注我们,获取更多相关资讯
翻译 by 左越晟、于妍、张保虎、王雪宝、宁太鑫、郑彦东
作为人体循环系统的动力中枢,心脏无时无刻不在跳动,是维系各个器官正常生理功能的“生命之泵”。身为“劳模器官”的心脏,经常面临衰老以及与之伴生的心血管疾病。在当前人口老龄化形式日趋严峻的情况下,寻找导致心脏细胞衰老的分子机制,并针对性的开发新型治疗手段和药物显得尤为迫切。哈佛大学的Jessica C. Garbern团队于2022年4月在Cardiovascular Research上发表了题为“Senescence mechanisms and targets in the heart”的综述文章,总结了心脏细胞衰老的分子特征及其诱导因素,揭示了心脏中各种细胞类型的衰老是心血管疾病发生的诱因,并阐述了延缓心脏细胞衰老及治疗心血管疾病的研究现状及未来方向。
摘要
细胞衰老是一种与机体衰老相关的不可逆的细胞周期停滞状态。心脏中不同细胞类型的衰老可以诱发诸如动脉粥样硬化、心肌梗塞和心脏纤维化等心血管疾病(CVDs)的发生。尽管与年龄相关的端粒缩短是复制性衰老的主要诱因,但氧化应激、代谢功能紊乱和表观遗传的改变等因素也可以促使机体进入衰老状态。因此,我们有必要了解细胞衰老的影响以及引发细胞衰老的分子机制,以便开发应对心血管疾病的新的治疗手段。本文中,作者总结了细胞衰老的分子机制,介绍了不同类型心脏细胞的衰老导致心血管疾病的原因,包括心肌细胞、心脏内皮细胞、心脏成纤维细胞、血管平滑肌细胞和瓣膜间质细胞等,并讨论了针对细胞衰老的分子机制的潜在治疗干预方案,以预防或治疗心血管疾病。

关键词:细胞衰老;个体衰老;心血管疾病;衰老细胞清除治疗
1
介 绍
细胞衰老的主要特征是由老化、DNA损伤或活性氧(ROS)水平升高等应激源诱导的不可逆的细胞周期停滞。不同类型心脏细胞的衰老与多种心血管疾病的发生相关,包括动脉粥样硬化、瓣膜性心脏病、心肌病和心律失常。心脏中细胞衰老的发生和发展与心脏疾病的进程和严重程度密切相关。了解细胞衰老诱导心血管疾病的机制对于相关治疗手段的开发至关重要。在本篇综述中,作者介绍了心脏中细胞衰老的机制,讨论了与不同类型心脏细胞衰老相关的疾病表型,以及针对这些疾病的潜在治疗方法。
2
细胞衰老的特征
细胞衰老是一种依赖于细胞类型和环境的异质性表型,定义为增殖能力的不可逆丧失。衰老可进一步分为几种亚型,包括复制性衰老、致癌基因诱导的衰老和应激诱导的过早衰老。衰老不同于静息状态(quiescence),静息状态是一种对营养信号变化的适应性反应,属于可逆的细胞周期阻滞,而细胞衰老的显著特征是细胞周期不可逆状态,停滞在G1/S期或G2期,导致其永久退出具有潜在增殖能力的细胞库。
鉴于其异质性表型,衰老细胞的鉴定具有挑战性。心脏和其他部位的衰老细胞有几个表征衰老的关键特征(图一)。衰老细胞会上调p53/p21、p16/视网膜母细胞瘤蛋白(Rb)通路和与DNA损伤反应(DDR)激活相关的标志物(p38丝裂原活化蛋白激酶(MAPK),磷酸化组蛋白2AX(γH2AX))。一些细胞显示与衰老相关的异染色质灶,因此增殖相关基因因不同的异染色质改变而被沉默。衰老细胞会表现出衰老相关分泌表型(SASP),其中可溶性信号因子、蛋白酶和不可溶性蛋白/细胞外基质(ECM)成分被分泌到周围细胞环境。SASP在维持衰老状态的同时也通过旁分泌效应影响周围细胞环境。衰老相关β-半乳糖苷酶(SA-β-Gal)活性通常在衰老过程中增加,但在静息态细胞中也可以增加。未来的工作仍需确定细胞衰老的直接标志物。
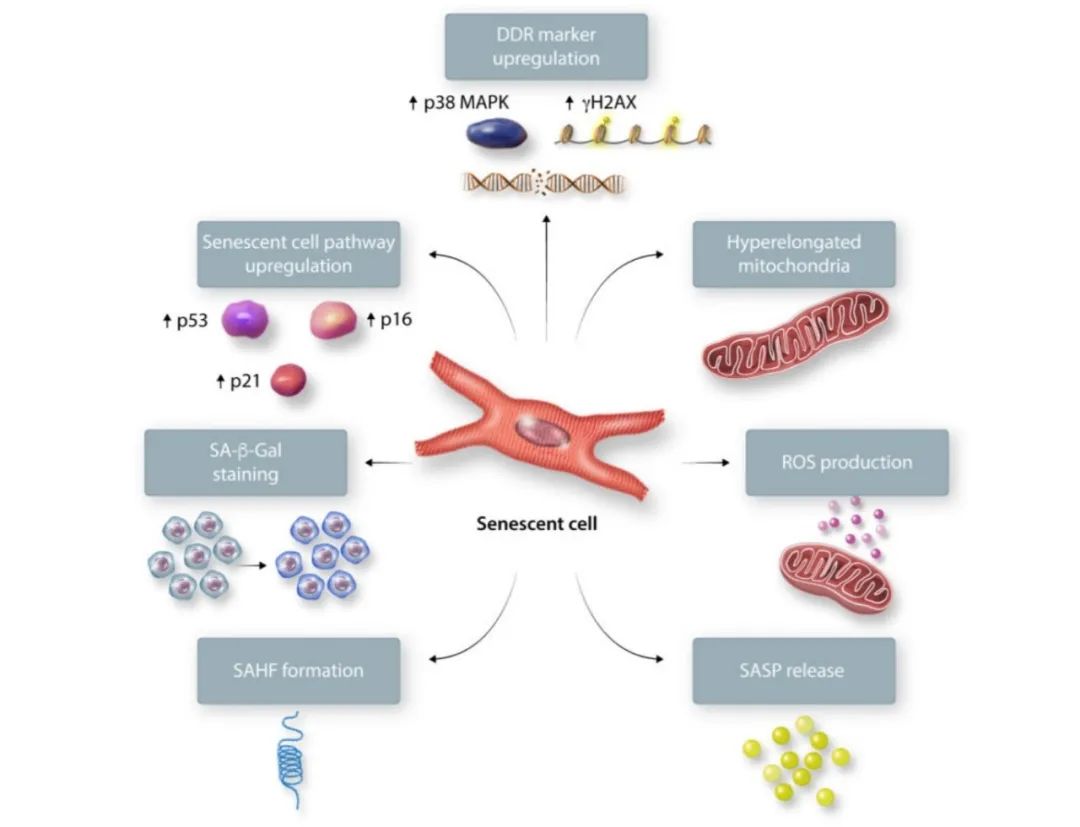
图一. 衰老细胞的标志物。衰老细胞表现出不可逆的细胞周期阻滞,p53、p21和p16升高,DNA损伤反应标志物p38 MAPK和γH2AX升高,线粒体过度延长,活性氧升高,SA-β-Gal活性、衰老相关异染色质灶(SAHF)和衰老相关分泌表型(SASP)增加。
值得注意的是,虽然细胞衰老可以加剧或加速衰老相关疾病,但衰老和凋亡途径之间的相互作用也可以在癌症预防中发挥关键的保护作用,并且在发育过程中也起着重要作用。应激源的剂量可能会影响这种细胞衰老结果途径的选择,如低剂量的化疗药物阿霉素可诱导衰老,高剂量则诱导新生大鼠心肌细胞凋亡。致癌基因的刺激可以通过上调p53或p16诱导某些细胞类型衰老,以防止其向肿瘤状态过渡。在成纤维细胞中,p53信号通路诱导的衰老能够将细胞的死亡途径从凋亡转移到坏死,这可能有益于抗癌治疗。此外,细胞衰老的程序性激活可能是正常胚胎发育所必需的,并可以与凋亡途径一起控制正常的形态发生。虽然SASP通常与炎症和肿瘤发生等负面影响相关,但SASP对伤口愈合、组织再生和免疫反应也有有益作用。
3
细胞衰老的分子调节剂
在这一部分中,作者广泛讨论了可能发生在不同类型细胞中衰老过程的分子机制(图二),在第4节中则讨论了这些过程如何介导心脏中不同细胞类型的衰老。
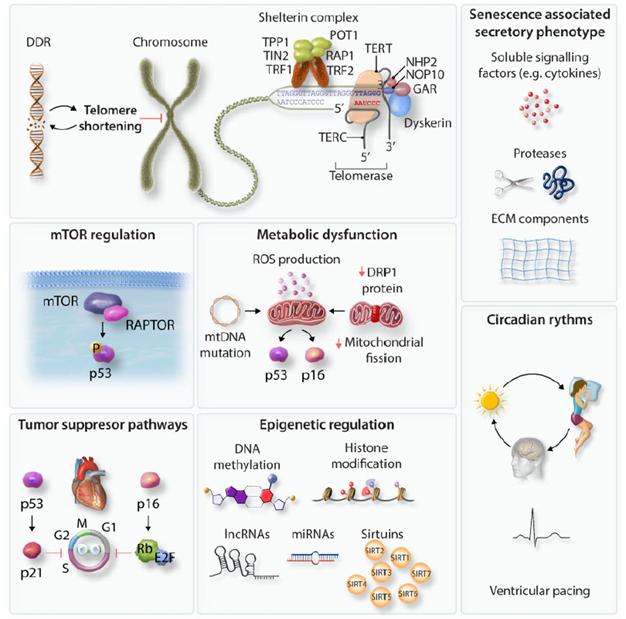
图二. 细胞衰老的分子调节剂。驱动细胞衰老的机制包括DNA损伤反应/端粒缩短、mTOR激活、代谢功能障碍、昼夜节律失调、SASP、肿瘤抑制途径和表观遗传的变化。
3.1 端粒和DNA损伤反应
端粒是串联重复序列,它覆盖了染色体末端,以保护染色体免受降解和防止两条染色体末端融合在一起。Shelterin环状复合物识别端粒结构,以防止DDR机制识别和处理端粒DNA。端粒随着细胞周期的重复而缩短,当达到一个临界的端粒长度时,Shelterin蛋白不再被招募来保护DNA环,从而激活DDR系统并启动细胞周期抑制。特别是在非心肌细胞中,端粒缩短随着衰老自然发生,进而引发细胞的复制性衰老。
3.2 调节衰老的肿瘤抑制蛋白
肿瘤抑制因子通过激活p53/p21和/或p16/Rb通路诱导细胞衰老发生,从而导致细胞周期阻滞(图三)。p53是一种转录因子,调节与代谢、自噬、DDR、细胞周期和凋亡相关基因的表达。p53的活性受到多种翻译后修饰的影响,包括泛素化、磷酸化和乙酰化。p53正向调控p21,而p21是周期蛋白依赖性激酶(CDK)抑制剂中的一员,也是p53在G1/S或G2/M期介导细胞周期阻滞所必需的。p21通过与半胱天冬酶结合抑制细胞凋亡,促进衰老状态。p16/Rb通路受到p16的抑制活性调控,p16结合CDK4/CDK6从而防止Rb磷酸化,导致细胞周期阻滞在G1/S期。p16/Rb通路也通过有丝分裂信号级联参与了ROS诱导,激活蛋白激酶C delta,从而创建一个正反馈循环维持衰老。
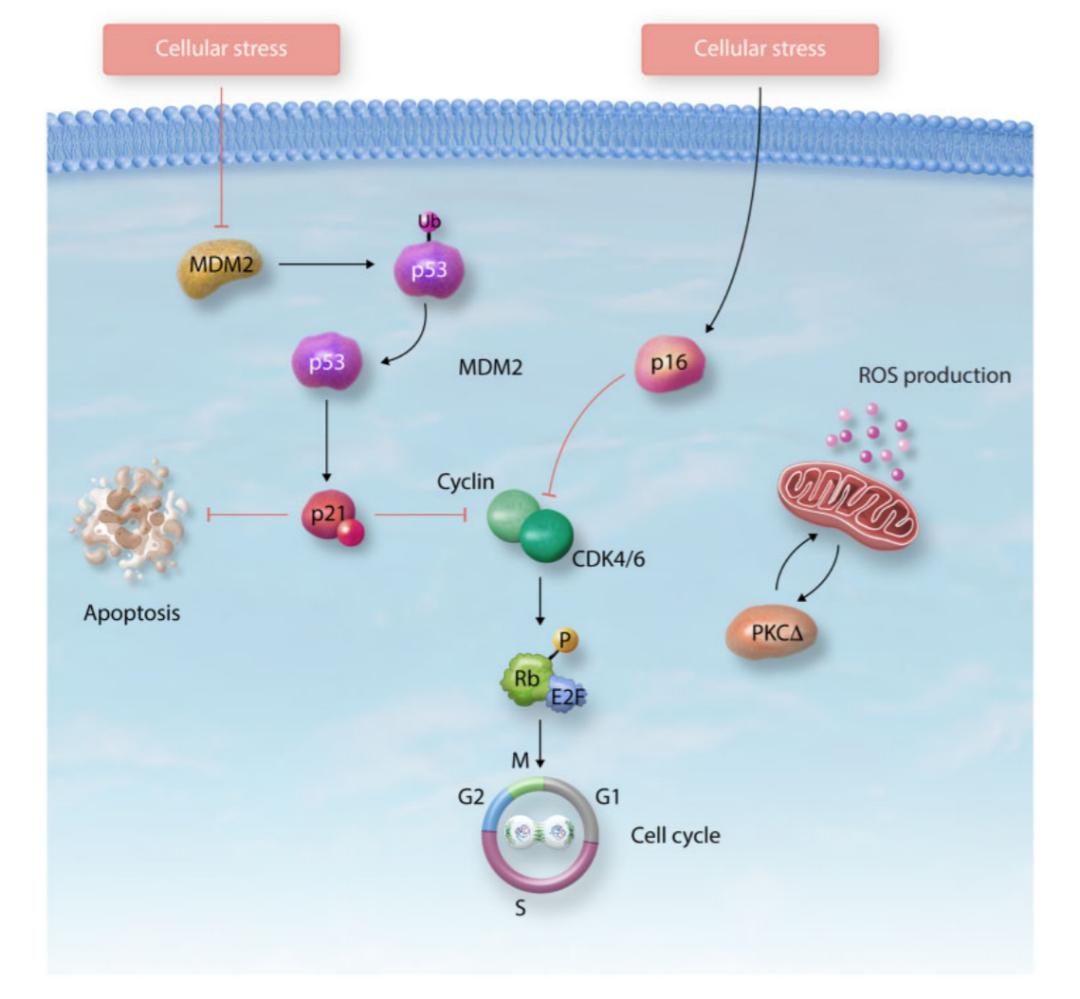
图三. 肿瘤抑制因子调控细胞衰老。p53/p21和p16/Rb通路通过细胞周期阻滞、ROS的产生和DNA损伤反应的激活在衰老过程中发挥核心作用。
3.3 雷帕霉素的机制靶点
雷帕霉素靶蛋白(mTOR)是一种丝氨酸/苏氨酸激酶,它响应各种环境和细胞内的信号来调节生长和代谢。mTOR可通过增殖调节因子(磷酸肌醇3-激酶(PI3K)/蛋白激酶B(AKT))的激活和致癌过程中衰老信号(p53)的正向调节来诱发衰老。mTOR控制着衰老和静息状态之间的抉择:随着p53和mTOR激活,细胞发生衰老;随着p53激活和mTOR抑制,细胞则变成静息状态。
3.4 线粒体动力学和功能障碍
由于线粒体裂变和融合蛋白之间的平衡被破坏,衰老细胞的线粒体过度延长。线粒体裂变蛋白FIS130或动力相关蛋白1(DRP1)水平的相对降低或线粒体融合蛋白、线粒体融合素1和2(MFN1/2)或OPA1水平的升高能够促进过度延长线粒体的衰老表型。线粒体融合是维持正常线粒体和心脏功能所必需的,而裂变则有助于通过线粒体吞噬去除功能失调、去极化的线粒体。在衰老过程中,裂变和融合过程的失衡可导致功能失调的线粒体和氧化蛋白的积累,这可能加剧衰老表型。线粒体功能失调与ROS增加有关,可导致包括巯基氧化、脂质过氧化和线粒体DNA(mtDNA)突变在内的氧化损伤增加。线粒体动力学异常导致的代谢紊乱是引发心肌细胞衰老的重要因素。
3.5 心脏衰老的表观遗传调控
表观遗传变化包括DNA甲基化、组蛋白乙酰化、染色质重塑和非编码RNA。与增殖细胞相比,衰老细胞保留了非常不同的甲基化特征,比如DNMT1调控导致晚期复制基因区域的低甲基化和特定启动子近端区域的高甲基化。这些效应被假定为可导致细胞周期抑制和随后的增殖阻滞/细胞周期退出。
组蛋白修饰在物理上改变染色质结构,同时招募各种含有组蛋白结合结构域的接头蛋白/效应蛋白对染色质进一步重构。Sirtuins(SIRT)是一个烟酰胺腺嘌呤二核苷酸(NAD+)依赖的组蛋白去乙酰化酶家族,可以防止多种细胞类型产生衰老表型。在心肌细胞中,SIRT1可以通过组蛋白的去乙酰化抑制心肌细胞中SASP的转录,而在内皮细胞中,SIRT1通过调节内皮一氧化氮合酶(eNOS)维持内皮细胞功能以减少氧化损伤。
非编码RNA(微小RNA(mi-RNA),长链非编码RNA(lncRNA))可以调节衰老和心血管疾病的进展。例如,当miRNA-22在衰老心脏中上调时,就会促进心脏成纤维细胞的衰老和迁移。此外,miRNA-29通过转化生长因子(TGF-β)/SMAD信号通路负调控H4K20me3,在心脏衰老过程中维持衰老状态。先前的综述描述了其他miRNAs在衰老和心脏损伤过程中的作用。lncRNA在心脏再生和发育中发挥重要作用,主要通过三种机制:与核糖核蛋白结合,结合和抑制miRNA(生成lncRNA-miRNA-靶点轴)以及与DNA片段结合形成核结构域。例如,在衰老心脏中过表达lncRNA H19可以通过抑制miR-19a激活p53/p21衰老驱动通路,促进心肌细胞衰老。此外,lncRNA SNHG12通过与DNA依赖蛋白激酶(DNA-PK)结合,作为DDR的调节剂调节其活性。敲除lncRNA SNHG12可导致DNA损伤修复受损和血管衰老,从而加快动脉粥样硬化。因此,lncRNA可能是衰老相关心血管疾病的潜在治疗靶点。
3.6 衰老相关分泌表型
SASP包含三大类分泌因子:可溶性信号传导因子,蛋白酶和不溶性蛋白质/ECM组分。SASP可溶性信号传导家族中的主要细胞因子包括白细胞介素-6(IL-6)和IL-1蛋白。DDR独立于p53调控之外,可以直接调控IL-6,影响表达IL-6R细胞表面受体的邻近细胞。IL-1蛋白在衰老过程中同样上调,作为SASP表达的主要前馈机制,促进核因子kappa B(NF-κB)介导的炎症级联反应的转录激活。SASP蛋白酶包括基质金属蛋白酶(MMPs)和丝氨酸蛋白酶。SASP促进不同类型心脏细胞衰老的一大机制在于将衰老信号由非心肌细胞传递给心肌细胞(图四)。
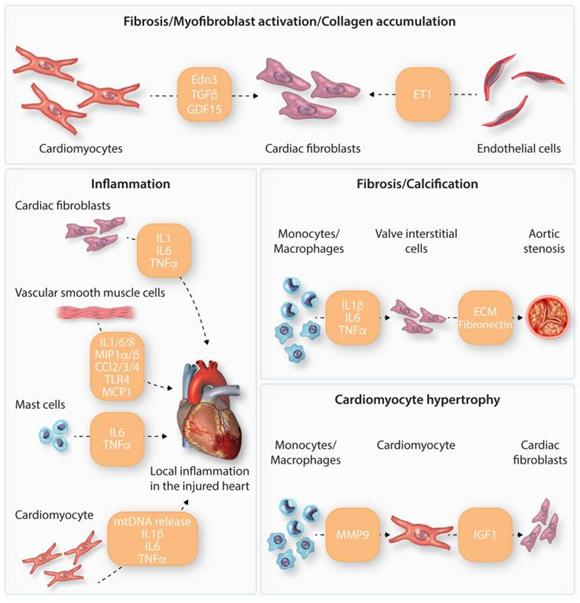
图四. 心脏中不同细胞类型的分泌组以及相关表型变化的示例。衰老心脏细胞分泌不同的衰老相关分泌表型相关因子,导致旁分泌信号传导和衰老表型恶化。这些分泌因子可导致纤维化,肌成纤维细胞活化,胶原蛋白合成,钙化,肥大和炎症等。
3.7 衰老的昼夜节律调节
昼夜节律可能在细胞衰老方面发挥作用,因为昼夜节律的失衡会缩短小鼠寿命并诱导免疫细胞衰老。在心脏中,昼夜节律相关基因Per2的突变可以通过AKT通路引起血管衰老。生物钟功能的失调可能在心血管疾病进程和心脏衰老方面具有不利影响,同时,血管平滑肌细胞(VSMC)的衰老会阻断昼夜节律信号的传递。
4
心血管疾病中的衰老心脏细胞
前文描述了不同类型心脏细胞在衰老过程中的表型变化,以及其如何影响心血管疾病的进展。虽然在某些情况下,全身性疾病可能先于细胞衰老,例如急性心肌梗死(MI),但仍有证据表明,在许多疾病中,细胞衰老是疾病发生的诱因。针对每种类型心脏细胞,我们列举了以下与细胞衰老相关的心血管疾病的例子。
4.1 心肌细胞
心肌细胞约占心脏总细胞数的30-40%,总细胞体积的80%。衰老的心肌细胞形态受损、起搏频率增加、收缩功能障碍、代谢功能障碍,分泌SASP因子以诱导邻近细胞(包括成纤维细胞)衰老。在心肌细胞衰老的不同机制中,代谢应激在诱导心肌细胞疾病表型中占主导地位。
4.1.1 心肌细胞衰老的分子机制
代谢功能紊乱可能对心脏中年龄相关功能的下降具有重要影响,因为影响能量产生的能量底物或应激源的变化可以导致心肌细胞衰老。具体来说,线粒体的动力学或质量控制的失调会导致ROS增加,进而造成氧化损伤增加。线粒体能够在几分钟内通过裂变或融合改变形态,以响应应激。此外,线粒体自噬(即去除受损线粒体以保护细胞免受整体代谢功能障碍影响的过程)失调可导致细胞衰老。具有心肌细胞特异性Drp1/Mfn1/Mfn2三基因缺失的小鼠比单独缺失其中一种基因的小鼠存活时间更长,这表明比起对线粒体动力学的整体破坏,裂变和融合过程的失衡危害更剧烈。
在心肌细胞中,通过FOXO3活化上调过氧化氢酶和超氧化物歧化酶2(SOD2),可以预防氧化损伤阻滞衰老。在线粒体中过表达过氧化氢酶可以保护心脏免受年龄相关代谢功能障碍的影响。随着年龄的增长,mtDNA突变会增加并导致ROS增加。从线粒体逃逸的mtDNA可以触发Toll样受体9(TLR9)信号通路,介导下游促炎细胞因子的释放,从而导致心肌细胞的炎症反应。这些炎症反应进一步维持SASP,并促进心血管疾病进展。这些证据表明,对于心脏中衰老相关的氧化应激治疗干预措施,特别是对于衰老心肌细胞相关的氧化应激来说,防止ROS的增加很重要。
此外,细胞周期调控因子还可以导致线粒体功能障碍,从而促进细胞衰老。胞质p53可以通过抑制Parkin蛋白介导的线粒体自噬来促进线粒体功能障碍。衰老过程中的p53活化会干扰糖酵解过程及心肌细胞中GLUT1/4依赖的葡萄糖转运,进而导致葡萄糖代谢受损。在患糖尿病的小鼠模型中,抑制p53可以预防细胞衰老和心肌病。细胞周期抑制因子Rb1和Meis同源盒2(Meis2)可以驱动衰老,急性心肌梗死后,Rb1和Meis2的抑制可以增强成年大鼠心肌细胞的增殖并减小梗死区域。
除了线粒体功能障碍外,心力衰竭期间心肌细胞会发生端粒缩短,DNA损伤。此外,表观遗传因素可影响心肌细胞衰老。例如,在组蛋白去甲基化酶KDM4D过表达的成体心肌细胞中,H3K9me3的丢失导致心肌细胞增殖,细胞周期基因表达增加,逆转细胞周期的退出。miRNA-34家族在衰老心脏中上调,导致心肌细胞收缩力降低。在衰老相关的心力衰竭中,miRNA-17-92家族在心肌细胞中特异性表达降低,导致纤维化重塑和功能下降。而适当的SIRT1过表达可防止心脏肥大、细胞凋亡、功能障碍和细胞衰老。SIRT7还与p53相互作用以介导p53脱乙酰,从而防止心肌细胞凋亡,具有潜在的延缓衰老的作用。
衰老心肌细胞的分泌组包括炎症细胞因子IL-1和IL-6、肿瘤坏死因子α(TNF-α)、内皮素3(Edn3)、生长和分化因子15(GDF15)及TGF-β。炎症标志物(IL-1、IL-6和TNF-α)在周围心脏微环境中诱导局部炎症,Edn3、GDF15和TGF-β被证明在心脏成纤维细胞中诱导纤维化和肌纤维母细胞活化。综上所述,与心肌细胞相关的SASP对周围的心脏细胞群具有广泛的旁分泌作用。
4.1.2 心肌细胞衰老的临床表征
衰老心肌细胞伴有收缩力受损及传导模式异常,通常会导致心肌病或心律失常。例如在杜氏肌营养不良症(DMD)模型中,抗肌萎缩蛋白缺失的小鼠的心肌细胞表现出衰老表型。此外,核纤层蛋白A(LMNA)突变与扩张型心肌病有关,可能与开放染色质区域增加导致核膜破裂而造成的基因组不稳定性有关。此外,蒽环类药物(一类用于治疗癌症的化疗药物)可导致与心肌细胞衰老相关的扩张型心肌病表型。给予阿霉素药物的大鼠心肌细胞在6个月后显示出mtDNA损伤和衰老标志物的增加。
心肌细胞衰老也可能增加室性心律失常的风险。衰老大鼠心室心肌细胞ROS的增加会影响心肌细胞响应电起搏的能力,并导致心律失常风险的增加。此外,与无室性心律失常的急性心肌梗死的患者相比,发生室性心律失常的患者的心肌细胞伴有端粒功能障碍增加,提示了心肌细胞的衰老。TMEM43(跨膜蛋白43)的突变,这与心律失常性心肌病和心肌细胞的衰老有关。因此,心肌细胞衰老不仅可能导致内在心肌细胞的功能障碍,而且还可能通过旁分泌信号诱导其他心脏细胞类型的衰老。
4.2 内皮细胞
内皮细胞约占心脏中非心肌细胞的60%。内皮细胞通过分泌血管活性化合物和生长因子来调节血管舒张和血管张力,在动脉粥样硬化斑块,心力衰竭 (尤其是舒张功能不全)和房颤的心脏中都可以观察到衰老的内皮细胞。
4.2.1 心脏内皮细胞衰老的分子机制
内皮细胞衰老可由氧化应激或血管炎症引发。代谢因素是导致内皮细胞衰老的一大原因,比如在以高脂肪或高碳水化合物饮食为主的西方人群中,观察到高尿酸血症或肾素-血管紧张素系统失调会导致内皮细胞衰老。通过miRNA或组蛋白乙酰化的表观遗传调节也可导致内皮细胞衰老。miR-217下调eNOS导致内皮细胞功能障碍并加剧APOE敲除小鼠的动脉粥样硬化,miR-217还可以通过SIRT1的下调加剧衰老表型。在内皮细胞中,SIRT1去乙酰化p53以干扰复制性衰老途径。
衰老内皮细胞导致内皮素-1(ET-1)增加,一氧化氮产生减少,这些都导致衰老内皮细胞与周围心脏细胞群之间的显著串扰。包括正反馈回路中的血管炎症和血管舒张受损,其中衰老内皮细胞的积累导致血管功能障碍,反之亦然。ET-1的分泌导致衰老性心脏成纤维细胞中胶原蛋白合成增加,与年龄相关的心脏肥大的胶原和纤连蛋白发生积累。在治疗方面,靶向衰老内皮细胞群仍然是避免与年龄相关的血管功能障碍的有效方案。当用神经调节素-1(NRG1)治疗时,通过ErbB酪氨酸激酶受体途径,内皮衰老表型减少。同样,端粒酶的表达也抑制了衰老相关的内皮功能障碍。通过延缓内皮细胞衰老可以避免在其他心脏细胞群中诱导衰老的下游效应。
4.2.2 心脏内皮细胞衰老的临床表征
内皮细胞衰老导致血管舒张受损和血管功能障碍,进而导致动脉粥样硬化、射血分数保留型心力衰竭(HFpEF)或肺动脉高压相关的心力衰竭等疾病。SIRT6缺乏或miR-217过表达可促进内皮细胞衰老,导致动脉粥样硬化。射血分数保留型心力衰竭风险随着年龄的增长而增加,并且与内皮细胞衰老和心肌纤维化有关。衰老加速模型小鼠(通过AKR/J小鼠的选择性近亲繁殖自然形成的过早衰老的SAMP8/TaHsd小鼠)在24周龄前出现舒张功能障碍,左心室肥厚和间质纤维化,其内皮细胞表现出p53乙酰化和SA-β-gal活性的上升。
涉及主-肺动脉分流问题的先天性心脏缺陷会导致肺动脉压升高,如果不及时治疗,可导致慢性肺动脉高压。肺动脉高压可以在年轻时通过主动脉、肺动脉分流的手术矫正来逆转,相反,晚期矫正则会导致肺脉管系统不可逆的损伤,并发展为慢性肺动脉高压(即艾森曼格综合征)。肺血管系统重塑能力的丧失与肺内皮细胞的衰老表型有关。
随着年龄的增长,房颤风险逐渐增加,并可能导致与血栓或心力衰竭相关的疾病和死亡。房颤与内皮细胞和成纤维细胞衰老有关,eNOS随房颤下调,这与内皮细胞功能障碍有关。凝血酶等循环因子可通过血管紧张素II途径诱导猪心房内皮细胞的细胞衰老,从而引发促炎、促纤维化和促血栓反应。更重要的是,衰老标志物p53和p16的表达水平与房颤的严重程度呈正相关。
4.3 心脏成纤维细胞
心脏成纤维细胞占心脏中非心肌细胞群的20%,其不仅能调节胞外基质的重塑,同时还能调控心脏微环境中的旁分泌通讯。心脏成纤维细胞的衰老有益于心肌梗死后的慢性伤口愈合,或阻碍衰老过程中的心肌纤维化。
4.3.1 心脏成纤维细胞衰老的分子机制
心脏成纤维细胞通过表达整合素和基质金属蛋白酶来维持胞外基质粘附所需的结构以及完整性。在急性心肌损伤后,激活的p53/p21通路会诱导心脏成纤维细胞的衰老。SASP蛋白之一的细胞通讯网络因子1 (CCN1)在心肌损伤后诱导成纤维细胞衰老来抑制心肌纤维化,表明成纤维细胞的衰老在某些情况下是有益的。与之相反的NEIL3是一种DNA糖基化酶,通过去除氧化的碱基对来最大限度地减少DNA的氧化损伤,而Neil3敲除小鼠的MMP2表达增加,促进了胞外基质降解并导致心脏破裂。
心脏成纤维细胞还通过旁分泌信号通路来调节心肌细胞的增殖、肥大性生长和衰老。在心脏损伤期间,炎性细胞因子(IL-1、IL-6)和TNF-α以及促纤维化因子TGF-β在心脏成纤维细胞中分泌增多。此外,心脏成纤维细胞产生的胰岛素样生长因子-1(IGF-1)也可以促进胶原蛋白合成和心肌细胞肥大。
4.3.2 心脏成纤维细胞衰老的临床表征
急性心肌损伤后,激活的心脏成纤维细胞从促炎状态转变为抗炎状态,从而促进瘢痕形成。心脏成纤维细胞的过早衰老会限制胶原蛋白的表达,这可能会阻碍伤口愈合早期的修复性纤维化,但也可以防止慢性伤口愈合期间的过度纤维化。成纤维细胞促衰基因的失活会导致心脏功能障碍和纤维化增加,过表达CCN1来诱导心脏成纤维细胞衰老则会减少纤维化和改善心脏功能。成纤维细胞衰老的有益影响需要与有害影响保持动态的平衡,比如在缺血/再灌注损伤期间,用navitoclax清除衰老细胞可以改善预后。Neil3敲除小鼠的心脏成纤维细胞过度增殖,但心脏破裂的风险也相应的增加:这是由于虽然心脏成纤维细胞在细胞周期中保持活跃,但Neil3缺失引起的氧化性DNA损伤会通过SASP介导的旁分泌信号通路促进胞外基质降解和心脏破裂,进而引发心脏微环境中的衰老表型。尽管心脏成纤维细胞的衰老会通过减少慢性伤口愈合中的纤维化产生有益影响,但非衰老成纤维细胞分泌的SASP因子(如MMP2)可能会对胞外基质重塑产生不利影响,从而增加心脏破裂的风险。
尽管急性损伤中成纤维细胞的过早衰老可以减少纤维化,但慢性病中的衰老表型则会加剧心脏纤维化。心肌纤维化程度随着年龄的增长而增加,并与HFpEF相关。miR-1468-3p促进心脏成纤维细胞的衰老,伴随p53和p16表达的上调、SA-β-gal活性的增加和TGF-β1信号通路介导的胶原沉积和心脏纤维化。SIRT6缺失还会通过TGF-β通路促进成纤维细胞转化为肌成纤维细胞,从而增加心肌纤维化。
4.4 血管平滑肌细胞
血管平滑肌细胞与内皮细胞协调血管功能以控制血压、血管张力和血流。衰老的血管平滑肌细胞会导致动脉粥样硬化和肺动脉高血压的形成与恶化。
4.4.1 血管平滑肌细胞衰老的分子机制
端粒缩短、DNA损伤、氧化应激和自噬功能障碍均可诱导血管平滑肌细胞衰老。衰老的血管平滑肌细胞中SA-β-Gal活性和p16/p21/Rb蛋白转录上调。核纤层前体蛋白A向核纤层蛋白A转换的过程紊乱导致核纤层功能失调,使细胞DNA更容易损伤。核纤层蛋白A基因突变导致的核纤层前体蛋白A上调或HIV蛋白酶抑制剂均通过降低ZMPSTE24(一种处理核纤层前体蛋白A的金属蛋白酶)的表达来促进血管平滑肌细胞衰老。持续的DNA损伤通过促进核纤层前体蛋白A的积累来激活成骨因子的分泌,进而导致了血管平滑肌细胞的矿化和随后血管的钙化。SIRT6缺乏也会因为组蛋白3在K9和K27(H3K9和H3K27)位点的高度乙酰化导致端粒DNA损伤从而促进血管平滑肌细胞的衰老。
衰老的血管平滑肌细胞中炎性细胞因子表达上调。IL-6和CC基序趋化因子配体2(CCL2)在老年血管平滑肌细胞中增加,TLR4介导的信号通路也是如此。血管平滑肌细胞的SASP表达量上升,包括单核细胞趋化蛋白1(MCP1)、巨噬细胞炎症蛋白-1α/β(MIP1α/β)、CCL3/4和IL-1/IL-6/IL-8,往往还会伴随抗炎细胞因子的下调。IL-1α表达导致邻近细胞的SASP激活和IL-6分泌增强,表明衰老血管平滑肌细胞的旁分泌功能会诱导产生心脏局部微环境的炎症。
4.4.2 血管平滑肌细胞衰老的临床表征
与内皮细胞一样,血管平滑肌细胞的衰老也会导致包括动脉粥样硬化和肺动脉高压在内的血管疾病。与正常内侧血管平滑肌细胞相比,动脉粥样硬化中的斑块血管平滑肌细胞伴随端粒缩短、p16和p21的表达上调、SA-β-gal活性和氧化DNA损伤增加。而其分泌的MCP1、MIP1α/β和CCL3/4会招募单核细胞、巨噬细胞和淋巴细胞,进一步加速斑块生长和增加破裂风险。斑块血管平滑肌细胞中SIRT6表达下调,而SIRT6过表达可阻止血管平滑肌细胞衰老。血管平滑肌细胞中核纤层前体蛋白A的积累诱导表现HGPS(Hutchinson-Gilford Progeria Syndrome,儿童早衰症)的临床表征,这类患者往往会患有严重的动脉粥样硬化,以及过早的衰老与死亡。此外,血管平滑肌细胞的衰老也会通过SASP促进产生肺动脉高压的病理特征。
4.5 瓣膜间质细胞
瓣膜间质细胞(VIC)是构成心脏瓣膜的主要细胞类型。瓣膜间质细胞的衰老也会导致瓣膜功能障碍,如主动脉瓣小叶钙化导致的主动脉瓣狭窄,其患病率随着年龄的增长而增加。钙化的主动脉瓣中p16和p53的表达上调,这表明衰老机制在疾病发生中起到重要作用。促炎性巨噬细胞分泌的TNF-α、IL-1β和IL-6在瓣膜间质细胞中短暂的诱导成骨表型,导致瓣膜纤维化和钙化。在体外同样可以通过TGF-β1通路在猪主动脉瓣膜间质细胞中激活MAPK来诱导衰老表型,从而增加ROS并形成钙化结节。钙化的主动脉瓣膜间质细胞中组蛋白3和组蛋白4高度乙酰化以及miR-214的下调会进一步诱导瓣膜间质细胞的成骨分化。粘液性二尖瓣疾病中的二尖瓣瓣膜间质细胞也会伴随miR-17、miR-20a、miR-30d和let-7c表达下调,p21表达和SA-β-gal活性增加,细胞活力降低和肌纤维母细胞分化。
4.6 免疫细胞与衰老心脏实质细胞的相互作用
免疫细胞在心脏衰老调节中的作用很复杂,在这里我们主要讨论免疫细胞如何调节心脏实质细胞的衰老,而不是免疫细胞本身的衰老。免疫细胞似乎具有双重作用——它们在消除衰老心脏实质细胞的同时也会通过炎症过程促进衰老。例如中性粒细胞在心肌损伤后介导巨噬细胞极化为抗炎表型;然而急性炎症性中性粒细胞也会对心肌损伤产生不利影响。心脏巨噬细胞通过去除衰老或受损的心脏细胞以维持心脏稳态。常驻心脏巨噬细胞分泌的MMP9等因子也会随衰老而增多,这反过来会促进心肌细胞肥大和胞外基质稳态失调,以及产生成纤维细胞衰老表征之一的胶原蛋白沉淀。急性心肌损伤后,调节性CD4+ T细胞会分泌促进伤口愈合和心肌细胞增殖的细胞因子。然而,随着衰老,T细胞也会导致心肌功能障碍,例如与缺乏CD4+ T细胞的老年小鼠相比,野生型老年小鼠的心脏收缩功能更差,这可能是TNF等炎症细胞因子水平较高导致的。肥大细胞在心肌损伤后会释放类胰蛋白酶,通过抑制肌钙蛋白I和肌球蛋白结合蛋白的过度磷酸化来调节蛋白激酶A的活性以保持心脏功能。但是在糖尿病性心肌病中,肥大细胞通过分泌TNF-α和IL-6来激活心脏成纤维细胞的TGF-β信号通路以促进胶原合成和心肌细胞凋亡,从而产生不利影响。深入研究如何利用免疫细胞的有益作用可能会为减缓心脏衰老提供新的治疗策略。
5
建立心脏衰老的动物模型
作者以前已经系统综述过可用于研究心脏衰老以及年龄相关心血管疾病的加速衰老小鼠模型。Lmna-/-小鼠有线粒体功能障碍和心肌病,并在6-8周龄时死亡,而Zmpste24(一种促进核纤层蛋白A加工的金属肽酶)缺失的小鼠,有传导异常(心动过缓、QRS波群延长),但心脏功能正常。缺失Cu/Zn-超氧化物歧化酶(Sod1)的小鼠更容易受到ROS增加导致的缺血-再灌注损伤。BubR1是胚胎发育过程必需的有丝分裂相关蛋白,缺失BubR1基因的小鼠会由于肌肉萎缩与心律失常而早逝。此外,AKR/J小鼠是由京都大学选择性近交所得的衰老加速小鼠(SAMP)模型,具有早衰与老化表征。对这些心脏衰老小鼠模型的研究可以帮助我们进一步阐明衰老心肌细胞群的生理变化及其在加重年龄相关心血管疾病之中的作用。
6
当下挑战,靶向治疗,未来方向
6.1 预防化疗后衰老
包括阿霉素在内的蒽环类药物可以在癌症治疗后引起心脏中多种细胞衰老,诱发心衰。阿霉素诱导新生鼠心肌细胞mtDNA损伤,上调p16的表达,在阿霉素处理鼠的左心室内还可见SA-β-gal的上调。阿霉素诱导的VSMCs衰老降低了SIRT1的表达,并降低了AMP活化激酶(AMPK)的激活,导致细胞呈促炎状态。泼尼松龙可以预防阿霉素处理后的VSMC衰老。LncRNA-MALAT1阻止miRNA-92a-3p对自噬调节蛋白酶4a(ATG4a)的抑制;ATG4a具有抗衰老及提升阿霉素处理后心肌细胞线粒体功能的作用。抑制化疗过程的细胞衰老也许可以削弱蒽环霉素处理心肌细胞的后续毒性影响。
6.2 衰老治疗
衰老细胞伴随糖酵解增加,DDRs增加,并释放SASP因子,基于这一特征,通过给药物添加蛋白靶标,可以达到移除衰老细胞(senolytics),抑制衰老表型(senostatics)的目的。在此,作者强调了一些已经在心血管疾病模型中测试过的能够尽量减少衰老细胞的影响的策略(图五)。
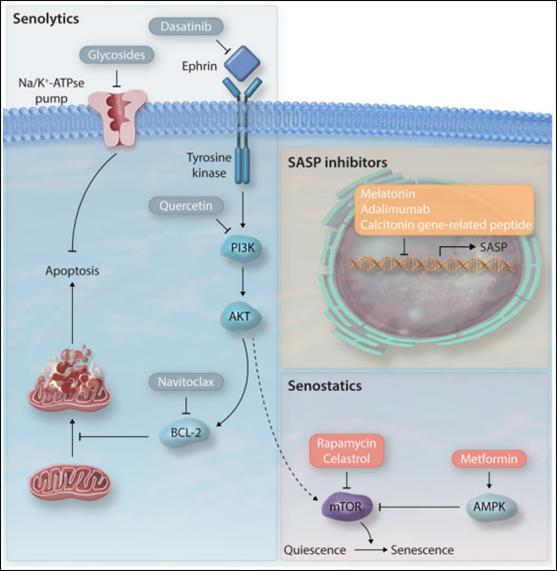
图五. 衰老治疗药物。衰老治疗药物旨在减少衰老细胞(senolytics),预防衰老状态(senostatics),延缓衰老进程(衰老相关分泌表型抑制因子)。
6.2.1 达沙替尼和槲皮素
达沙替尼和槲皮素是在筛选已知促凋亡药物时最早被发现的两种清除衰老细胞的药物。达沙替尼是一类酪氨酸激酶抑制剂,通过阻止Ephrin配体与受体间的相互作用诱导衰老细胞凋亡。槲皮素是一种抑制PI3K的黄酮醇,增加SIRT1的表达,抑制mTOR信号通路。槲皮素可降低ApoE-/-小鼠主动脉的脂质沉积,增加SIRT1表达。槲皮素与达沙替尼联合使用可以清除电离辐射暴露后小鼠体内p16表达的衰老细胞,并改善老年鼠左心室收缩功能。同样的,槲皮素和达沙替尼联合治疗可以降低高脂血症小鼠衰老标志物表达,改善血管功能,减少主动脉钙化。
6.2.2 ABT263
ABT263会抑制抗凋亡蛋白BCL-2家族的活性,导致肺内衰老成纤维细胞死亡;在小鼠模型中,ABT263可以促进心肌梗死后衰老心肌细胞的清除,提高生存率,降低纤维化;还可以改善左心室收缩功能,降低心脏纤维化与肥厚水平,提高Ang-II诱导心衰小鼠的传导速度。
6.2.3 强心苷
Na+-K+-ATP酶转运体利用ATP将Na+从细胞外带入,将K+从细胞内排出。强心苷(包括地高辛、乌本苷和海葱次苷A)是从洋地黄中提取到的一类化合物,可以抑制Na+-K+-ATP酶活性,导致细胞膜去极化,并使电化学梯度紊乱。衰老细胞比非衰老细胞的质膜去极化程度更高,对强心苷也更敏感。地高辛可以清除肺纤维化模型小鼠的成纤维细胞。
6.2.4 代谢调节剂
mTOR通路控制着静止态到衰老态的转换,当mTOR被抑制时,可以延长小鼠寿命。雷帕霉素可以促进儿童早衰症患者的成纤维细胞自噬,清除早衰蛋白,从而延缓衰老。雷公藤红素可抑制mTOR通路,增强血管平滑肌细胞自噬能力,降低细胞内ROS水平,阻止Ang-II诱导的衰老。二甲双胍是一种糖尿病常用药物,也被证实可通过AMPK通路激活从而延长寿命。当给ApoE-/-小鼠服用二甲双胍后,可抑制小鼠内皮细胞衰老及动脉粥样硬化斑块的形成。
6.2.5 抑制衰老相关分泌表型
衰老相关分泌表型因子多种多样,但是衰老相关分泌表型的抑制可以阻止下游炎症反应导致的细胞衰老加速。褪黑素可以通过聚ADP核糖聚合酶和CREB结合蛋白阻止H2BK120乙酰化,从而抑制人原发性胎儿肺成纤维细胞中及胚胎成纤维细胞衰老相关分泌表型,抑制SASP基因上调。降钙素基因相关肽可通过上调抗衰老蛋白klotho抑制心肌纤维母细胞衰老和衰老相关分泌表型。由于许多可以激活衰老相关分泌表型的通路(比如NF-κB, mTOR)对于其他生理过程(如肿瘤检测和免疫系统)的调控也是极其重要的,所以在不诱发不良反应的标准下抑制衰老相关分泌表型是极富挑战性的难题。
6.3 个性化治疗
精准治疗旨在基于疾病表型与分子机制为患者量身打造治疗措施。促进特定类型心肌细胞衰老的分子机制或可用于疾病的靶向预防和治疗。例如,DMD中缺失营养不良聚糖蛋白会促使衰老,原因可能在于核结构的失调以及基因组的不稳定。精准医疗已经在部分DMD的患者上开展,该类患者由于肌营养蛋白基因移码突变,通过反义寡核苷酸的传递绕过或跳过DNA突变区,导致翻译提前终止。作者先前也论述了如何将对疾病的表观组学、代谢组学、蛋白组学及微生物学机制的深入认知应用于特定心血管疾病表型。
6.4 细胞替代
细胞替代疗法是通过人干细胞分化来源的细胞来改善功能紊乱的细胞。由于心脏中心肌细胞增殖较弱,故以多能干细胞诱导心肌细胞以改善心衰患者心功能是有很大意义的。此外,将源于年轻小鼠的骨髓干细胞抗原1(Sca1)阳性细胞转移至年老小鼠体内,抑制了年老小鼠的衰老,促使心脏内皮细胞年轻化。最后,在细胞层面制定心脏年轻化策略时,还应当要考虑到免疫细胞与心脏常驻细胞之间内在交互作用的影响。
7
结 论
越来越多的研究表明,细胞衰老可能导致心血管系统疾病的病理生理变化。理解特定类型心脏细胞衰老在特定疾病表型中的作用可使我们发展出更优的靶向治疗策略以延缓或阻止心脏衰老相关疾病。当下治疗手段也正着眼于清除衰老细胞和抑制细胞衰老。从遗传学、表观遗传学、代谢组学的角度,深入探索心脏细胞的衰老机制,也许可以为心血管疾病患者提供更进一步的个性化治疗方案。

原标题:《【重磅综述】心脏衰老的分子机制及干预靶点》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2024 上海东方报业有限公司

