- +1
羧酸脱羧酰基化反应及机理研究
以下文章来源于CCSChemistry ,作者CCS Chemistry
CCSChemistry.
CCS Chemistry是由中国化学会创办的高水平旗舰新刊,面向全球科学家,收录化学各领域高质量原创科技论文。关注CCS Chemistry,即时获取期刊相关资讯。
酮作为最重要的羰基化合物之一,涉及了有机合成中的众多官能团合成和转化,因而开发绿色高效的合成方法成为有机化学工作者研究热点。南京大学谢劲、朱成建团队组利用光氧化还原/镍协同催化羧酸脱羧酰基化反应策略,实现了温和条件下利用羧酸和羧酸硫酯高效构筑非对称烷基酮和芳基-烷基酮。
目前合成酮的途径主要依赖于高活性试剂和消耗化学计量的还原剂,这使得酮分子的化学选择性和多样性合成成为一个巨大的挑战。鉴于商品化羧酸储量丰富且结构多样,开发从羧酸合成酮的新方法吸引了诸多研究人员的兴趣。这类反应的例子包括Friedel-Crafts酰化,有机金属试剂对酰胺的亲核加成和过渡金属催化的交叉偶联反应。近年来,多个课题组已经报道了用酰胺、酸酐或活化的2-吡啶基硫酯作为酰基化试剂的光氧化还原/金属催化的C(sp3)-H酰化反应的方法。在这些反应中,杂原子相邻的C(sp3)-H或简单烷烃C-H通过HAT过程,形成自由基,从而控制区域选择性。南京大学研究团队围绕着羧酸直接构建醛、酮方面已开展了系列工作(Nat. Commun. 2018, 9, 3517;Angew. Chem. Int. Ed. 2019, 58, 312;Nat. Commun. 2020, 11, 3312)。然而,仍然需要开发一种实用的方案,使用一种现成的化学试剂作为烷基自由基源,目的是在没有冗余的保护和脱保护过程的情况下构建复杂的酮。

图1 两种羧酸合成酮
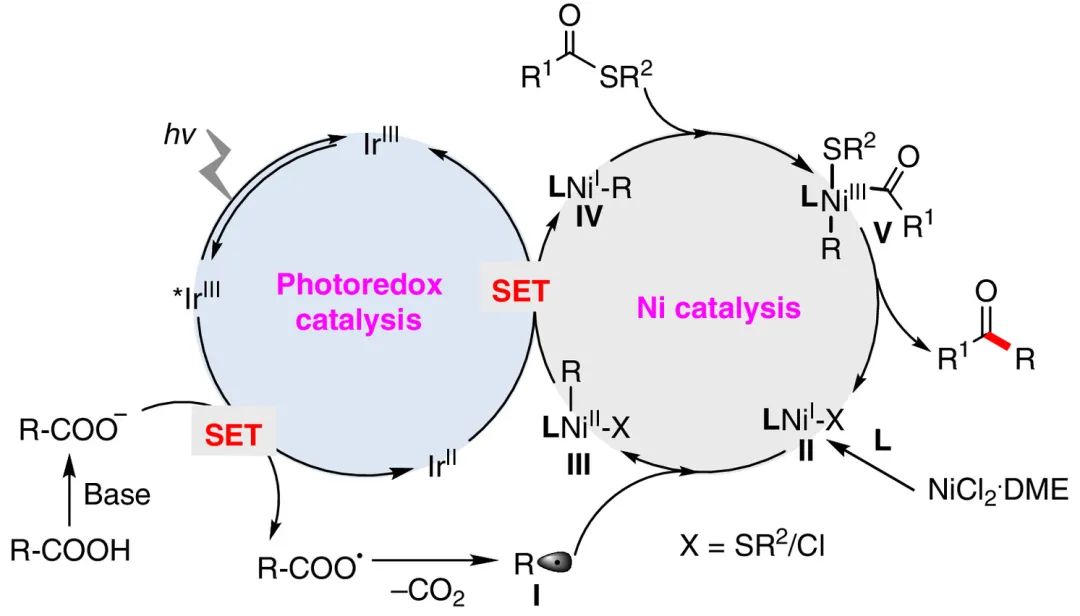
图2 反应机理图
在生物体内,羧酸可以通过辅酶A(CoA)形成硫酯键的活化模式,进行酰基传递、扩链,从而实现酶催化的脱羧酰基化反应(图1a)。受此启发,作者期望将过渡金属催化和光催化结合,前者负责活化硫酯形成活性酰基源,后者经历单电子氧化羧酸脱羧形成自由基(图1b),进而实现温和条件下脱羧酰化合成多样化的酮结构。近年来,可见光介导的自由基脱羧是形成C-C或C-X键的有效策略,例如脱羧炔基化、烯基化、烷基化、芳基化,氟化和硼化,然而,到目前为止还没有关于脱羧酰化的报道。
基于这些假设,图2中提出了一种合理的机制。最初,三价光敏剂铱Ir[dF(CF3)ppy]2(dtbbpy)PF6的经蓝光辐照产生激发态的铱物种。羧酸盐阴离子可与激发态光催化剂(E1/2red[*IrIII/IrII] = +1.21 V vs SCE)发生单电子转移(SET),生成烷基自由基(I)并释放CO2。在含氮配体的镍催化剂前体可还原为LNiX物种(II),其与生成的烷基(I)进行自由基加成以生成二价烷基Ni物种(III)。这可以接受来自Ir(II)物种的额外电子,以生成高活性的一价烷基镍R-Ni(I)物种(IV)。随后硫酯的C-S键与R-Ni(I)物种的氧化加成形成Ni(III)物种(V)。最后,还原消除将形成的C-C键,完成催化循环。
在优选的反应条件下,作者考察了可通过该酰化反应构建芳基烷基酮的市售羧酸的范围(图3)。许多苯环上带有富电子和贫电子官能团的羧酸都能在光/镍协同催化的体系中顺利脱羧,完成酰基化,以中等至优异产率(最高98%)合成相应的酮(3a–3y)。值得注意的是,含有芳基溴(3k)和活性N–H基团(3q和3r)的羧酸底物,在传统金属催化的交叉偶联反应中面临官能团兼容问题,而在此体系中被很好的保留。一些药物分子和生物分子如阿克他利(3v,73%)、吲哚美辛(3x,77%)、瑞格列奈前体(3w,49%)和唑吡坦前体(3y,82%)也可用于该反应。此外,含N的环状羧酸,包括脯氨酸(3cc)、氮杂环丁烷酸(3dd)等也以40-60%的产率生成相应的芳烷基酮。但是,三级羧酸不反应(3mm)。一般来说,直接脱羧的产生烷基自由基可确保良好的区域选择性并补充已报道的C-H酰基化方案。
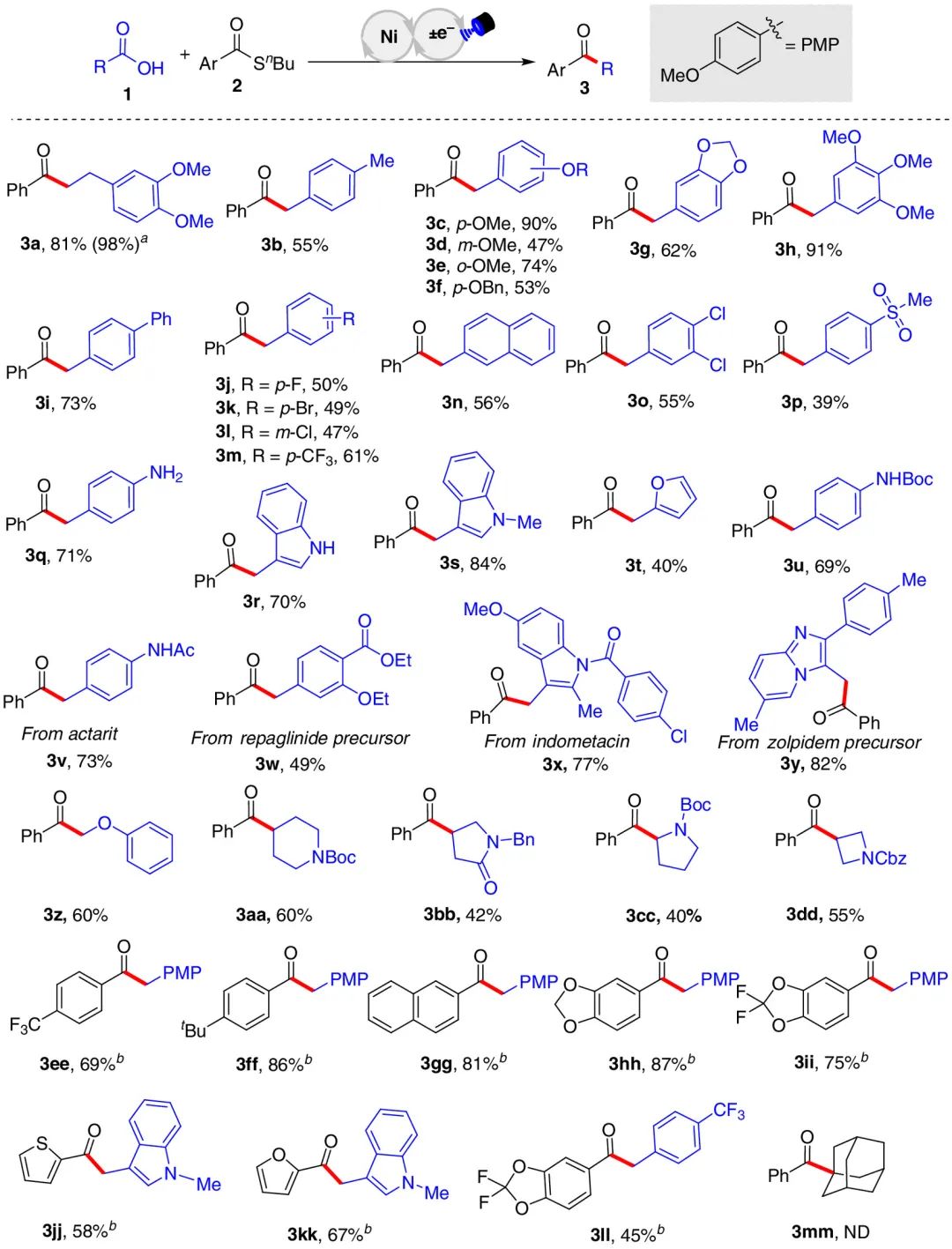
图3 羧酸的底物范围
作者评估了作为酰基供体的硫酯的多样性。如图4所示,苯环上的富电子基团和缺电子基团对C–S键的插入几乎没有影响,并以中等至良好的产率转化成酮(3ee–3ll)。芳基羧酸硫酯和烷基羧酸的硫酯都能作为出色的酰基供体,以中等至优异的产率合成了非对称烷基酮(4a-4p)。

图4 硫酯的底物范围
这种光氧化还原/镍催化反应的高效率和良好的官能团耐受性,在合成复杂酮的后期应用方面也有出色的表现(图5)。一些药物分子,如酮洛芬(5a)、洛索洛芬(5b)、奥沙普嗪(5c)和吲哚美辛(5f,5h),在标准条件下以中等产率选择性地转化成相应的酮。有趣的是,使用复杂脂肪酸和相应的硫酯时,能够构建更为复杂的二烷基酮。甚至利用羧酸原位形成硫酯,还能实现两种羧酸一锅法的克级制备非对称二烷基酮。
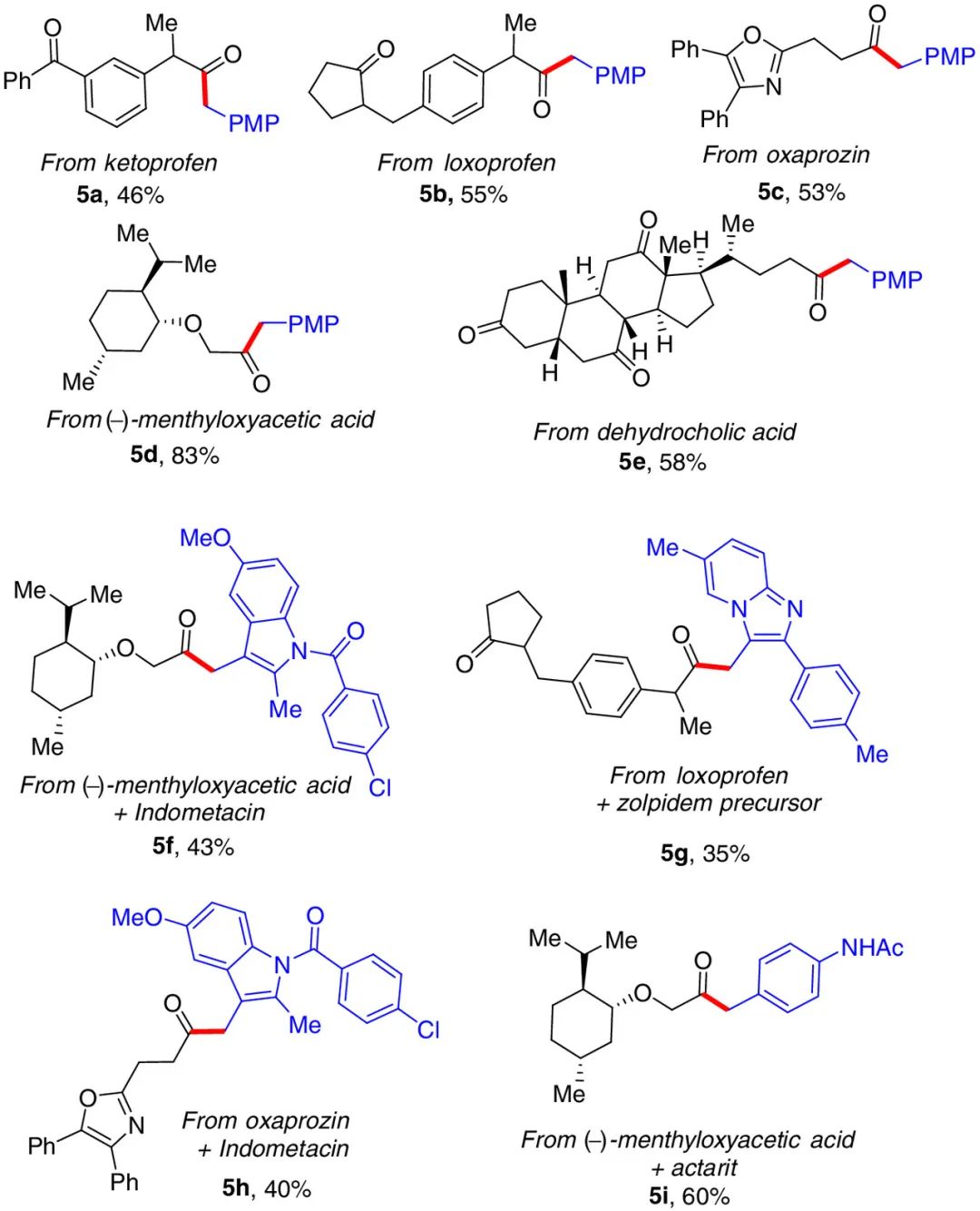
图5 复杂酮分子的合成
作者通过循环伏安实验(CV),自由基捕捉实验,电子顺磁共振实验(EPR),荧光淬灭实验以及对照的控制实验,并结合密度泛函理论(DFT)计算,推测可能的反应机理。以苯甲酸正丁硫醇酯和对甲氧基苯乙酸为模板,进行苄基自由基9的酰基化反应的DFT计算,提出了两种主要的反应路径(图6)。在路径1中,Ni(I)-Cl与SET引发并与苯甲酸硫酯1a络合形成Ni(0)物种1A,该过程的自由能变化在光催化循环内为12.1 kcal/mol (SET1,图6)。中间体1A继续与1a进行氧化加成以生成Ni(II)物种1B,能垒仅为3.5 kcal/mol。1B随后通过过渡态1B-TS 与自由基9结合,并形成中间体1C。反应继续进行,还原消除1C,得到目标产物3a和一价Ni(I)-SBu。最后,活性催化剂通过-SnBu和Cl-之间的配体交换再生,自由能为1.4 kcal/mol。路径1的决速步是从Ni(I)-Cl到1A-TS,总势垒为15.6 kcal/mol。路径2,从Ni(I)-Cl与9的无能垒自由基加成开始,形成中间体2A。Cl-阴离子通过Ir(II)与2A分离,形成Ni(I)物种2B。2B与1a结合,然后通过2C和2C-TS进行氧化加成,生成Ni(III)中间物种2D,其比路径1中的1C低7.9 kcal/mol。Ni(III)中间物种2D的能量也低于2A,因此路径2的决速步是2A至2B,能垒为10.3 kcal/mol。由于路径1的总能垒高于路径2,因此路径2在理论上似乎更可行。

图6 苄基自由基的酰基化DFT计算
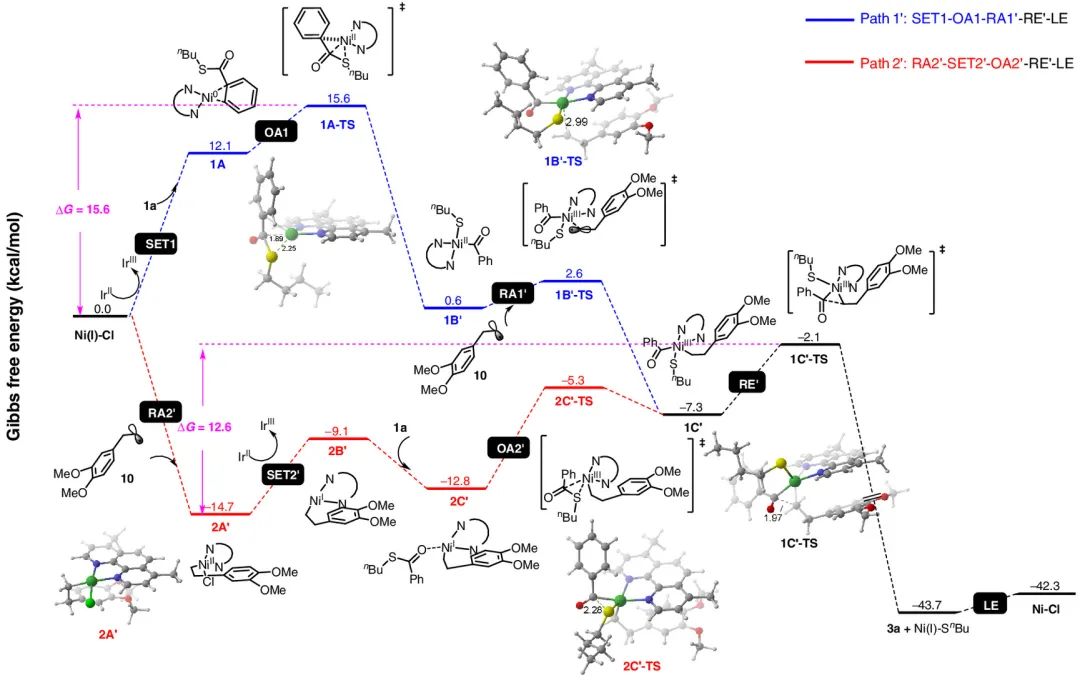
图7 烷基自由基的酰基化DFT计算
对于链状羧酸的脱羧酰基化过程(图7),路径2’的总能垒(12.6 kcal/mol)也是低于路径1’(15.6 kcal/mol),因此,证明了一开始的机理假设,在交叉偶联中镍催化剂经历了由一价镍为起始的Ni(I)/Ni(II)/Ni(I)/Ni(III)/Ni(I)催化循环。
综上所述,该研究工作发展了第一个光氧化还原/镍协同催化羧酸与芳香族和脂肪族硫酯的脱羧酰化反应。该反应可以在温和条件下利用商品化的羧酸为原料,构建结构多样的芳烷基酮和非对称二烷基酮,具有良好的化学选择性和官能团兼容性,代表了羧酸脱羧官能团化的重大进展。两种不同羧酸在克级规模的一锅法脱羧酰化反应和合成复杂酮的后期应用进一步提高了其合成效用。此项研究得到了国家自然科学基金、江苏省自然科学基金、中央高校基础研究基金、江苏省“创新创业人才计划”的资助。文章以Research Article 的形式发表在CCS Chemistry上,已在官网“Just Published”栏目上线。
文章详情:
Decarboxylative Acylation of Carboxylic Acids: Reaction Investigation and Mechanistic Study
Xiaopeng Wu†, Jie Han†, Siyu Xia, Weipeng Li,* Chengjian Zhu* and Jin Xie*Cite this:CCS Chem. 2021, 3, 2581–2592
文章链接:https://doi.org/10.31635/ccschem.021.202101197
扫码在线阅读
扫描或长按左侧二维码,
在线阅读全文
▼
CCS Chemistry
▼
CCS Chemistry是中国化学会独立出版的旗舰新刊,所有作者读者免费发表和阅读(Diamond Open-Access)。
CCS Chemistry网址:https://www.chinesechemsoc.org/journal/ccschem
Facebook:Chinese Chemical Society-CCS
Twitter: CCS Chemistry
扫描或长按以下二维码,关注CCS Chemistry微信公众号,及时了解CCS Chemistry发表的最新杰出研究成果。
中国化学会
Chemsoc
原标题:《羧酸脱羧酰基化反应及机理研究》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2024 上海东方报业有限公司

