- +1
【重磅综述】机体衰老与NAD+之间不得不说的故事
以下文章来源于老顽童说 ,作者老顽童说

老顽童说
公众号致力于传播衰老相关的前沿科研进展和趣味科普,帮助大家更深入地了解衰老背后的科学故事~

关注我们,获取更多相关资讯
翻译 By 刘黎啸、尹剑、蔺舫民、李明恒、孙国强
“青春须早为,岂能长少年”,忙碌奔跑只为达理想彼岸,为此,每一个细胞都在拼命奋战。小身体,大世界,在我们的身体中有这样一种物质,它存在于所有活细胞中,是生存和健康的发动机,也是维持人体年轻态的明星物质,它就是人体中的一种重要辅酶——烟酰胺腺嘌呤二核苷酸(NAD+)。多年来,我们发现了NAD+作为“发动机”的重要性,但再好的发动机也会有能源枯竭的一天,进而影响到与其相连的一系列配件,最终会影响整个工厂的正常运转,即人体发生的衰老过程。那么,NAD+究竟为何有这样的能力,又如何保持细胞活力而影响人体的衰老进程呢?巴克老龄化研究所(Buck Institute for Research on Aging)所长Eric Verdin教授带领团队于2020年12月22日在Nature Reviews Molecular Cell Biology上发表的综述文章“NAD+ metabolism and its roles in cellular processes during ageing”为我们总结了当前已知的“真相”,解析了NAD+和衰老及衰老相关疾病间千丝万缕的联系,并为相关研究道路树立了指示牌,提示着前方的困难和方向。
通讯作者:Eric Verdin (图源网络)
摘要
烟酰胺腺嘌呤二核苷酸(NAD+)作为氧化还原反应的辅酶,是能量代谢的中心分子。对于Sirtuins、CD38和多聚ADP-核糖聚合酶等非氧化还原的NAD+依赖酶来说,NAD+也是一个必要的辅助因子。NAD+可以直接或间接影响许多关键的细胞功能,包括代谢途径、DNA修复、染色质重塑、细胞衰老和免疫细胞功能。这些细胞过程及其发挥的功能对于维持组织和代谢稳态以及健康衰老至关重要。值得注意的是,在包括啮齿动物和人类在内的多种模式生物中,衰老伴随着组织和细胞NAD+水平的逐渐下降,NAD+水平的下降与许多衰老相关疾病存在因果关系,包括认知能力下降、癌症、代谢性疾病、肌少症和虚弱。许多与衰老相关的疾病可以通过恢复NAD+水平来减缓甚至逆转。因此,靶向NAD+代谢已成为一种潜在的改善衰老相关疾病、延长人类健康寿命和最大寿命的治疗方法。然而,NAD+如何影响人类健康和衰老尚需更多研究,包括更加深入地探究调节NAD+水平的分子机制,探究在衰老过程中有效恢复NAD+水平的方法及安全性,补充NAD+对衰老人群的有益影响等关键内容。
正文
烟酰胺腺嘌呤二核苷酸(NAD+)是氧化还原反应的重要辅酶,也是能量代谢的中心。对于去乙酰化酶(Sirtuins)和聚(ADP-核糖)聚合酶(PARPs)等非氧化还原的NAD+依赖酶,NAD+也是必不可少的辅助因子。
NAD+最初被发现是因其在酵母提取物中起到调节代谢速率的作用,后来又被证明是氧化还原反应中主要的氢化物受体。NAD+接受氢化物离子并形成还原态NADH的能力对于所有生命形式的代谢反应是至关重要的,并调节参与多种分解代谢途径的脱氢酶的活性,包括糖酵解、谷氨酰胺酵解和脂肪酸氧化。在真核生物中,这些反应中所接受的电子随后被传递至电子传递链,以形成ATP。NAD+也可以被磷酸化形成NADP+,而NADP+也可以作为氢化物受体形成NADPH,参与抗氧化应激和需要还原剂的合成代谢途径,如脂肪酸合成。
除了能量代谢,NAD+还是数百种酶的辅助因子或底物,因此在调节细胞过程和细胞功能方面具有多种作用,其中许多功能仍在研究中。NAD+水平与健康之间的联系几乎在一个世纪前就已建立。1937年,Conrad Elvehjem发现糙皮病(以皮炎、腹泻和痴呆为特征)是由饮食中缺乏烟酸引起的,导致低NAD+和NADP+水平。最近,人们发现低NAD+水平与多种疾病状态相关,包括代谢和神经退行性疾病,以及较低的NAD+水平与啮齿动物和人类的衰老相关。因此,人们对NAD+代谢如何影响疾病的起源,特别是与衰老相关的疾病有了新的兴趣。在这方面,用NAD+前体烟酰胺核苷(NR)和烟酰胺单核苷酸(NMN)恢复NAD+水平已成为一种重要的治疗衰老相关性疾病的方法,而且已经有证据表明在啮齿动物模型中能够产生有益的效果。
在这篇综述中,作者着重介绍了过去5年中NAD+领域的研究工作,尤其是NAD+前体和最终NAD+水平在衰老和疾病状态下影响生理和寿命的分子机制。由于这些机制是复杂的且受多因素调控,因此作者分为多个部分进行介绍。首先作者回顾了在衰老过程中调节NAD+合成和降解通路等方面的主要新发现。其次,作者讨论了低水平NAD+对衰老相关疾病重要分子过程的可能影响,包括DNA修复、表观遗传学和基因表达的调控、细胞代谢和氧化还原平衡的调节。接着,作者描述了衰老中的NAD+依赖机制,包括代谢紊乱、免疫系统失调、细胞衰老和神经退行性变。最后,作者回顾了大量近期高质量的临床前研究,这些研究探讨了如何恢复NAD+水平来治疗老年相关疾病,包括大量使用NAD+前体的研究,以及促进NAD+生物合成的小分子药物。在文章结尾,作者介绍了这些不同的策略和这些研究的结果,以概述基于调节NAD+水平延长人类健康寿命和寿命的治疗前景。
(一)细胞NAD+代谢
NAD+的亚细胞定位高度集中在胞浆、线粒体和细胞核。这些部位的NAD+是被独立调控的,与此一致的是,NAD+生物合成或降解所涉及的酶也分布在不同的位点。NAD+是多种代谢途径和细胞过程的关键代谢物和辅酶。首先,NAD+的还原需要维持能量平衡和细胞的氧化还原状态。NAD+也被两种消耗NAD+的酶不断地逆转:NAD+糖水解酶,也被称为NADases (CD38、CD157和SARM1),和去乙酰化酶家族的Sirtuins 和 PARPs,均具有多种重要的细胞功能。它们利用NAD+作为底物或辅助因子,并产生副产物烟酰胺(NAM)(图1)。
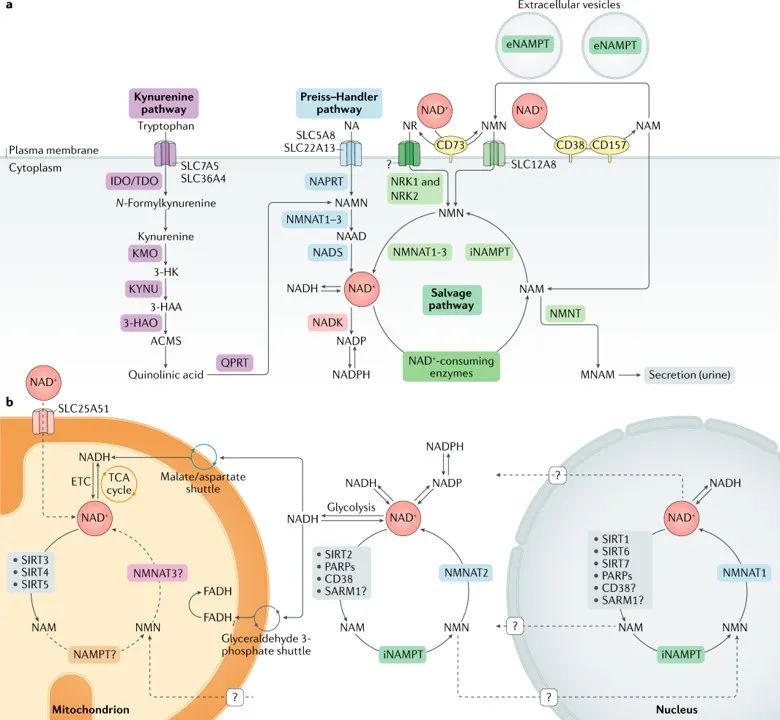
图1 | NAD+代谢。 a | 烟酰胺腺嘌呤二核苷酸(NAD+)生物合成途径。NAD+水平由3个独立的生物合成途径维持。犬尿酸途径(或从头合成途径)利用食物中的氨基酸色氨酸产生NAD+。色氨酸通过转运蛋白SLC7A5和SLC36A4进入细胞。在细胞内,色氨酸被限速酶吲哚胺2,3-双加氧酶(IDO)或限速酶色氨酸2,3-双加氧酶(TDO)转化为N-甲酰基脲嘌呤。N-甲酰基犬尿素转化为l-犬尿素,再经犬尿素 3-单加氧酶(KMO)转化为3-羟基犬尿素 (3-HK),再经色氨酸2,3-双加氧酶(KYNU)转化为3-羟基氨基苯甲酸(3-HAA)。利用3-羟基苯甲酸加氧酶(3HAO)合成α-氨基-β-羧酸ε-半醛(ACMS)。该化合物可自发缩合重组为喹啉酸,喹啉酸经喹啉酸磷酸核糖基转移酶(QPRT)转化为烟酰胺单核苷酸(NAMN),并与Preiss-Handler通路汇合。Preiss-Handler通路使用饮食烟酸(NA),NA通过SLC5A8或SLC22A13转运蛋白进入细胞,通过烟酸转磷酸核糖基酶 (NAPRT)生成NAMN,NAMN然后通过烟酰胺单核苷酸腺苷酸转移酶(NMNAT1 NMNAT2和NMNAT3)转换成烟酸腺嘌呤二核苷酸(NAAD)。这一过程是通过NAD+合成酶(NADS)将NAAD转化为NAD+完成的。NAD+回收途径回收作为NAD+消耗酶(乙酰化酶, 聚(ADP -核糖)聚合酶(PARPs),NAD+糖水解酶和环ADP -核糖合成酶CD38, CD157和SARM1)酶活性副产物的烟酰胺(NAM)。首先,细胞内的烟酰胺磷酸核糖基转移酶(iNAMPT)将NAM循环为烟酰胺单核苷酸(NMN),NMN再通过不同的NMNATs转化为NAD+。NAM可被烟酰胺N -甲基转移酶(NNMT)选择性甲基化,并通过尿液分泌。在细胞外空间,NAM是胞外酶CD38和CD157的副产物,可以通过胞外NAMPT (eNAMPT)转化为NMN。然后NMN被CD73去磷酸化成为烟酰胺核苷(NR),NR通过一个未知的核苷转运体转运到细胞中。NMN可以通过NMN特异性转运体(小肠中SLC12A8)导入细胞。在细胞内,NR通过烟酰胺核苷激酶1和2 (NRK1和NRK2)形成NMN。然后NMN被NMNAT1,NMNAT2和NMNAT3转换成NAD+。b | 不同亚细胞室的NAD+代谢。NAD+稳态是不同亚细胞间室合成、消耗和再生的平衡,受亚细胞特异性NAD+消耗酶、亚细胞转运蛋白和氧化还原反应的调控。NAD+前体通过三种生物合成途径进入细胞(a部分)。在细胞质中,NAM通过细胞内NAMPT (iNAMPT)转化为NMN。然后NMN被NMNAT2转化为NAD+,NMNAT2是该酶的胞质特异性亚型。NAD+在糖酵解过程中被利用,产生NADH, NADH通过苹果酸/天冬氨酸穿梭和甘油醛3-磷酸穿梭转移到线粒体基质。通过苹果酸/天冬氨酸穿梭进入的线粒体NADH被电子传递链(ETC)中的络合物I氧化,而从甘油醛3-磷酸穿梭中生成的还原黄素腺嘌呤二核苷酸(FADH2)被络合物II氧化。最近,人们发现了哺乳动物NAD+线粒体转运体SLC25A51,并证实它与细胞器中完整的NAD+摄取有关。线粒体中NAD+的回收途径尚未完全解决,但已经提出了一种特异的NMNAT亚型(NMNAT3)的作用。在线粒体中,NAD+被依赖NAD+的线粒体SIRT3-SIRT5消耗,产生NAM。目前还不清楚在线粒体内NAM是否能被转化回NMN或转化为NAD+,其他前体是否能通过线粒体膜运输来促进NAD+的合成。核NAD+池可能通过核孔扩散与胞质NAD+池相平衡;然而,完整的动态在很大程度上仍未被探索。一种核特异性NMNAT亚型(NMNAT1)已被描述,它是核NAM回收NAD+途径的一部分。MNAM, N1-甲基烟酰胺;TCA,三羧酸。
框1| 通过NAM回收途径生成NAD+
烟酰胺(NAM)回收途径由前体NAM或上游NAD+维生素前体烟酰胺核苷(NR)或烟酰胺单核苷酸(NMN)生成烟酰胺腺嘌呤二核苷酸(NAD+),这些前体物质可通过牛奶、水果、蔬菜和肉类等多种日程食品来摄入补充。由于NAD+不具有细胞通透性,因此认为除NMN外,所有的膳食前体,包括烟酸、NAM、NR和色氨酸,都是直接输入到细胞中用于NAD+的生物合成。支持这一观点的是,在导入NMN之前,细胞外的NMN必须被5' -核苷酸酶CD73转化为NR,从而在细胞内通过烟酰胺核苷激酶1 (NRK1)转为NMN。最近的一项研究显示,存在一种特异的NMN转运蛋白SLC12A8在小肠中高表达,这表明NMN是一个直接进入NAD+生物合成途径的切入点。需要进一步的研究来确定这个转运蛋白与疾病的生理相关性,NMN和其他NAD+前体摄取的动力学和机制,以及它们在哺乳动物的每个组织和/或细胞中的选择性表达和作用。
通过NAM挽救途径回收NAD+是在不同种类的NAD+消耗酶(包括糖水解酶(CD38、CD157和SARM1)、蛋白质去乙酰化酶(Sirtuins)和聚(ADP-核糖)聚合酶(PARPs))介导的不可逆降解后恢复NAD+水平的基本步骤。虽然每一种酶作用于NAD+活性和用途不同,但所有消耗NAD+的酶都作为NAD+降解的副产物产生NAM;这是由NAM回收途径酶烟酰胺磷酸核糖基转移酶(NAMPT)转化为NMN。NMN也可以通过NRK1和NRK2从NR中生成,并在回收路径的最后一步由烟酰胺单核苷酸腺苷转移酶NMNAT1、NMNAT2和NMNAT3转化为NAD+。NMNAT的三种亚型有不同的亚细胞定位(NMNAT1在细胞核;NMNAT2在高尔基体细胞质面;NMNAT3在线粒体),且被报道在各自的细胞室调节NAD+水平,但也影响其他细胞内NAD+存储。
NAMPT广泛表达,主要在依赖高NAD+水平的过程中上调,如免疫细胞激活和遗传毒性应激。通过调节乙酰化酶和PARP的活性,NAMPT介导的NAD+生物合成影响细胞代谢和对炎症、氧化和遗传毒性应激的反应。此外,NAMPT的表达受SIRT1反馈回路中的生物钟机制(CLOCK-BMAL1)调控,这与体内NAD+水平的昼夜振荡有关。哺乳动物NAMPT以两种形式存在:细胞内NAMPT (iNAMPT)和细胞外NAMPT (eNAMPT)。作为一种NAD+生物合成酶,NAMPT形成二聚体。许多细胞类型产生eNAMPT,其分泌在脂肪组织和癌细胞中分别受到SIRT1或SIRT6介导的去乙酰化的积极调控。最近研究表明,在小鼠和人的血液循环中,eNAMPT携带在细胞外囊泡中,它负责增强原发性下丘脑神经元细胞内NAD+的生物合成。除了在调节NAM回收中的作用,eNAMPT先前被鉴定为一种推测的细胞因子(也被称为前B细胞集群增强因子(PBEF))。多种免疫和代谢疾病患者血清中eNAMPT单体水平升高。然而,目前还不清楚NAMPT的酶活性对其细胞因子样功能是否必要。因此,区分eNAMPT的单体和二聚体形式可能是解决目前有关eNAMPT酶活性和功能争论的一个途径。总的来说,这些研究表明,在远端细胞中,eNAMPT以自分泌的方式维持NAD+,而且,值得注意的是,它也作为一种内分泌信号影响远端组织中的NAD+水平和炎症反应。
框2| NNMT和NADK介导NAD+的分解代谢
烟酰胺N -甲基转移酶(NNMT)是一种利用S -甲基腺苷(SAM)作为甲基供体将烟酰胺(NAM)的环氮甲基化,将其转化为N1 -甲基烟酰胺(MNAM)的酶。MNAM可通过醛氧化酶进一步氧化,产生N1 -甲基-2-吡啶-5-甲酰胺和N1 -甲基-4-吡啶-3-甲酰胺,并随MNAM一起分泌在尿中。NNMT将NAM转化为MNAM,有效地将从NAM回收途径得到的烟酰胺腺嘌呤二核苷酸(NAD+)中循环回NAM,并影响整体NAD+水平。因此,人们对NNMT在疾病状态(如肥胖、癌症和老龄化)中作为NAD+水平关键调控因子的作用越来越感兴趣。例如,在肥胖期间,内脏白色脂肪组织和肝脏中NNMT表达增加,而且似乎大多有负面影响。这包括甲基供体SAM和NAD+在白色脂肪组织和肝脏对高脂肪饮食的反应的消耗。因此,提高NNMT活性导致参与纤维化的基因启动子区甲基化减少,以及代谢和炎症基因的异常表达。因此,NNMT似乎是一种关键的代谢酶,通过调节生物活性分子(如NAD+和SAM的消耗)的水平,将NAD+的代谢与基因表达的控制联系起来,从而产生MNAM。
NNMT在调节NAD+中的作用提示NNMT在衰老过程中起重要作用。秀丽隐杆线虫NNMT的同源基因ANMT-1调控MNAM的产生可支持这一观点。在蠕虫体内,MNAM作为醛氧化酶GAD-3的底物,GAD-3产生过氧化氢,反过来,过氧化氢又作为促进长寿的激效信号。缺乏ANMT-1的蠕虫不再受益于依赖sir-2.1的寿命延长。这些结果表明,通过NNMT摄入NAD+至少对线虫的寿命有延长作用。然而,目前尚不清楚NNMT的激活在哺乳动物衰老过程中起什么作用,尽管NNMT的表达和活性似乎在啮齿类动物衰老过程中增加。例如,作者最近发现衰老小鼠的肝细胞中NNMT基因表达增加,在喂食高蛋白饮食的老年小鼠的肝细胞中也观察到类似的表达增加。此外,用NNMT抑制剂治疗老年小鼠可以激活衰老的肌肉干细胞并促进衰老期间的肌肉再生。因此,与线虫不同的是,在哺乳动物中,早期证据表明NNMT可能促进与衰老相关的疾病。因此,靶向NNMT可能为治疗老年相关疾病提供一种可行的治疗途径。
NAD+的另一种代谢命运是NAD+激酶(NADK)直接磷酸化产生NADP(H), NADP是调节细胞内氧化还原平衡和合成代谢过程(如脂肪生成)的主要还原力来源。最近一项使用体外同位素示踪标记纳入NADP(H)的研究表明,NADK占NAD+消耗的10% 。然而,总NADP+池比NAD+池少20倍。此外,在如老龄化等NAD+水平下降的情况下,NADP+水平也会下降。因此,NADP+水平似乎与NAD+水平直接相关,并随升随降,这使得NADK不太可能是主要的NAD+池。然而,最近研究表明,NADK是AKT激酶的直接靶点,可磷酸化丝氨酸44、丝氨酸46和丝氨酸48残基,从而增强NADK271的激活。因此,鉴于AKT信号在衰老过程中异常增加,NADK异常激活可能是衰老过程中NAD+水平下降的部分原因。因此,需要进一步的研究来确定NADK在老年和老年相关疾病中调节NAD+和NADP+池的作用。
因此,NAD+介导多个重要的生物过程,且需求量一直很高。NAM可以通过NAM补救途径循环利用来维持NAD+水平(详见框1)。此外,一些主要在肝脏的细胞可以从多种膳食来源从头合成NAD+。因此,细胞内NAD+不断合成、分解和回收来维持细胞内NAD+水平的稳定(图1)。然而,在衰老过程中这种分解代谢和合成代谢过程之间的平衡转变,NAD+的降解速度超过了细胞产生NAD+的能力,也超过了细胞有效回收NAM的能力。此外,过量的NAM可能通过其他的代谢途径分解,有效地将其从NAM回收途径转移,进一步影响NAD+水平(详见框2)。NAD+糖水解酶、去乙酰化酶和PARPs除了作为消耗NAD+的酶外,在衰老和年龄相关疾病中也有不同的作用。虽然增强去乙酰化酶的激活已成为延长寿命和健康寿命的一种方式,但异常激活PARPs和NAD+糖水解酶,如CD38,可能发挥相反的作用,加剧衰老表型。
(1)NAD+合成通路
NAD+可以通过犬尿酸途径从左旋色氨酸中从头合成,也可以通过Preiss–Handler通路从维生素前体(如烟酸(NA))中合成(图1a)。除了NAD+,犬尿酸通路还利用左旋色氨酸产生犬尿喹啉酸、血清素和吡啶甲酸等生物活性分子。值得注意的是,从头合成途径对NAD+水平的贡献仍不清楚。除肝脏外,大多数细胞不表达通过犬尿酸通路将色氨酸转化为NAD+所需的全部酶。大多数色氨酸在肝脏代谢生成NAM,并被释放到血清,被外周细胞吸收,并通过NAM回收途径转化为NAD+。此外,在某些情况下,免疫细胞,如巨噬细胞,也会从色氨酸中产生NAD+。因此,除了肝脏之外,从头合成途径似乎是一种更间接的机制,有助于维持系统范围内的NAD+水平,大多数NAD+来自于NAM回收途径(框1)。
(2)NAD+消耗通路
通过去乙酰化酶的消耗。去乙酰化酶自发现以来受到了人们的广泛关注,参与调节关键的代谢过程、应激反应和衰老生物学进程。哺乳动物去乙酰化酶家族包含7个基因,编码7个不同亚细胞定位的蛋白(SIRT1和SIRT6在细胞核中,SIRT7在核仁,SIRT3、SIRT4和SIRT5在线粒体,SIRT1、SIRT2和SIRT5在胞浆)(图1b)。这些依赖于NAD+的酶的亚细胞定位突出了细胞内NAD+的局部波动,这些NAD+自身受到相应去乙酰化酶的调节,可能选择性地影响细胞器特异性的去乙酰化酶活性和细胞代谢。
去乙酰化酶在细胞中处于持续活跃的状态。在基础条件下,SIRT1和SIRT2大约占了NAD+总消耗量的三分之一。此外,NAD+水平的升高与禁食和热量限制期间的乙酰化酶激活密切相关。值得注意的是,乙酰化酶的活性与生物钟是耦合的,SIRT1和SIRT6调控核心时钟转录因子和下游昼夜节律相关的转录组的活性。此外,SIRT1和NAD+回收途径的关键酶—烟酰胺磷酸核糖转移酶(NAMPT)在NAD+水平的昼夜节律调控中发挥关键作用,NAMPT受生物钟的调控,生物钟提供了一个反馈回路,导致NAD+水平的节律性振荡(框1)。
早期在酵母中的研究证实了去乙酰化酶在基因沉默中的作用,在2000年,人们认为其具有NAD+依赖的去乙酰化酶的活性。去乙酰化酶活性最初报道主要包括去除目标蛋白质的赖氨酸残基的乙酰基,该反应包括两步:首先NAD+被裂解为NAM和ADP-核糖,其次目标蛋白质的乙酰基转移到ADP-核糖,最终生成中间产物—肽基- ADP-核糖。随后,乙酰- ADP -核糖被释放出来(图2a)。此外,去乙酰化酶家族的一些成员介导非乙酰赖氨酸酰化(如琥珀酰化、丙二酰化和脂肪酸酰化),但迄今为止对其细胞功能的了解还很有限。SIRT4和SIRT6也具有ADP -核糖基转移酶的功能,表明去乙酰化酶对细胞调控的作用机制不仅限于蛋白乙酰化的调控,还需要进一步的研究。
尽管检测亚细胞内NAD+水平和去乙酰化酶活性受到技术的限制,但随着技术的发展,我们现在对这些酶的不同细胞作用有了一些深入的了解。具体而言,最近的证据表明,SIRT1、SIRT6和SIRT7是DNA修复和基因组稳定性的关键调控因子,线粒体SIRT3、SIRT4和SIRT5和SIRT1以及核SIRT1调节线粒体稳态和代谢。与它通过过氧化物酶体增殖物激活受体-γ共激活因子1α去乙酰化促进线粒体生物发生的作用明显相反,SIRT1也涉及到线粒体自噬缺陷线粒体的转换。然而,在这两种情况下SIRT1似乎都是线粒体质量维持的关键因素。总的来说,去乙酰化酶已经成为理解和描述NAD+水平如何影响衰老的各种细胞过程中的细胞稳态的关键角色,提高其活性是旨在对抗衰老治疗的关键焦点。
通过PARPs的消耗。人类PARP蛋白家族由17种具有多聚(ADP-核糖基)聚合酶或单体(ADP-核糖基)聚合酶活性的蛋白组成。简单地说,PARP介导NAD+的分解产生副产物NAM和ADP-核糖,ADP-核糖作为单一或共价链接的聚合物又加入到PARP本身和其他受体蛋白中,这个过程称为聚(ADP-核糖)化(图2b)。在所有的PARP中,只有PARP1、PARP2和PARP3定位于细胞核,响应早期DNA损伤,在DNA损伤修复中起关键作用(图1b)。
PARP1是这一家族中最有特点的成员,至少在对DNA损伤做出响应时,它承担着大约90%的PARP活性。激活后,PARP1将自身与组蛋白和其他蛋白结合,充当支架,招募并激活其他DNA修复酶和蛋白到损伤部位,启动DNA修复。由于PARP1的高活性,DNA损伤与大量的NAD+消耗有关。PARP1作为一种NAD+应答信号分子,广泛与衰老过程相关。然而,PARP1是NAD+的主要消耗者之一,不仅存在于急性DNA损伤的细胞中,也存在于正常和其他病理生理条件下,表明PARP1对调节NAD+稳态的关键作用。例如在喂养高脂肪饮食的小鼠中,那些处理了PARP抑制剂或缺乏PARP1和PARP2的小鼠NAD+水平增加,SIRT1活性提高以及线粒体功能改善,并保护免受胰岛素抵抗。在着色性干皮病A组患者中,PARP1激活、NAD+水平下降和SIRT1活性抑制,且在早衰症、共济失调毛细血管扩张症和柯凯因氏综合征患者中都观察到了这一相关性。值得注意的是,用PARP1抑制剂或NAD+补充剂治疗柯凯因氏综合征老鼠促进寿命延长和改善PARP1超活化所造成的显著变化表型,有力证明了在相应DNA损伤和基因毒性压力时PARP1的激活导致下游的NAD+内稳态紊乱。值得注意的是,PARP1拮抗SIRT1活性的能力最有可能是因为这些酶定位于相同的细胞室(在这种情况下为细胞核),竞争相同的NAD+池。然而,与SIRT1相比,PARP1对于NAD+具有更低的Km和更高的Vmax,PARP1由于其更强的结合亲和力和更快的酶促动力学,很可能在NAD+分解上胜过SIRT1。
PARP2在结构上与PARP1相似。它们具有类似的细胞过程所需的催化结构域,包括DNA修复和转录调控,约占PARP活性的10%。PARP2活性也可能影响NAD+的生物利用度。与敲除PARP1的小鼠一样,敲除PARP2的小鼠的SIRT1活性增强,代谢功能改善,并保护其免受高脂肪饮食导致的肥胖的伤害。PARP3在DNA修复中也很重要,表明PARP1、PARP2和PARP3存在大量的重叠和潜在的冗余。其他PARPs(PARP4-PARP17)在细胞或器官内NAD+稳态和整体代谢中的功能尚未完全确定,但它们在调节细胞内NAD+水平方面的作用被认为不那么重要。总的来说,靶向PARPs,特别是PARP1,在衰老领域是一种很有前途的治疗策略。然而,需要更多的研究来充分了解PARPs对年龄相关NAD+水平下降的影响。
通过CD38和CD157引起的消耗。CD38和CD157是多功能胞外酶,具有糖水解酶和ADP -核糖环化酶活性。NAD+的糖水解是主要的催化反应,裂解NAD+内的糖苷键生成NAM和ADP -核糖,而ADP -核糖环化酶的活性生成环状ADP -核糖(图2c)。CD38还进行碱基交换反应,在酸性条件下将NAD(P)+的NAM交换为NA,生成烟酸腺嘌呤二核苷酸(磷酸盐)(NAAD(P))(图2c)。值得注意的是,环状ADP -核糖、NAAD(P)和ADP -核糖都是关键的Ca2+动员第二信使,说明了CD38在激活Ca2+信号和调节基本细胞过程(如免疫细胞激活、生存和代谢)中的关键作用。重要的是,除了NAD+和NADP+,NMN是CD38的替代底物,而CD157消耗NR作为替代底物(图2c)。因此,用小分子抑制剂靶向CD38和CD157可以使这些常用的NAD+前体代谢产物更有效地恢复衰老个体的NAD+水平。对于CD157酶在细胞生物学或衰老中的作用知之甚少。然而,最近的证据表明,与CD38一样,CD157在衰老组织中表达上调,并可能在衰老相关疾病(如风湿性关节炎和癌症)中发挥作用。
虽然CD38和CD157在基因上是同源的,都属于同一酶家族的成员,但它们的结构、定位和在疾病中的作用是不同的。CD38是一种跨膜蛋白,具有Ⅱ型取向和/或Ⅲ型取向,在20世纪70年代末被确定为T细胞激活标记物,但现在已知其广泛表达,特别是在炎症过程中。CD157是一种以糖磷脂酰肌醇为锚定的蛋白,首次在造血系统的骨髓腔室中被鉴定。然而,其他细胞也表达CD157,包括B细胞祖细胞、潘氏细胞和肠道、胰腺和肾脏的内皮细胞。
除了酶的功能外,CD38和CD157还扮演着细胞受体的角色。例如,CD38是一种与CD31相互作用的粘附受体,通过内皮介导免疫细胞的运输和外渗。CD38 - CD31轴似乎促进了慢性淋巴细胞性白血病中淋巴细胞的增殖反应,表明CD38在血癌中起着有害的作用。然而,对于CD38 - CD31相互作用的功能后果知之甚少。此外,CD38被认为通过抗菌功能介导免疫,考虑到CD38敲除小鼠的一个主要表型是它们无法增强对细菌的免疫应答。目前尚不清楚CD38的酶促功能对其抗菌功能是否必要,以及这些功能在多大程度上依赖于NAD+。然而,CD38在抗菌素耐药性中的作用可能与从需要NAD+生存和生长的细菌中隔离NAD+或NAD+相关代谢产物有关。
CD157作为受体的作用还没有完全研究。目前有几条证据表明,特异性单克隆抗体激活CD157可促进中性粒细胞和单核细胞的转运。此外,CD157与整合素相互作用,形成瘙痒反应基因1蛋白(SCRG1)识别的受体,促进人间充质干细胞的自我更新、迁移和成骨分化。然而,这些受体的功能是否与NAD+代谢有关还不清楚。
通过SARM1的消耗。直到最近,SARM1才与CD38和CD157一起被归为NAD+糖化酶和环化酶家族。SARM1依靠Toll/白细胞介素受体(TIR)结构域发挥其酶活性(图2c),该结构域在过去的报道中通常参与蛋白-蛋白相互作用,并未发现它的催化活性。SARM1是否对NAD+有调节作用乃至调节程度都尚不清楚。然而,SARM1介导的NAD+降解在轴索损伤后的轴索变性中起着关键作用。SARM1主要在神经元中表达,促进神经元形态发生和炎症反应,也可由巨噬细胞和T淋巴细胞等免疫细胞表达,并调节其功能。最初发现SARM1是Toll样受体信号下游,可以直接与TRIF (TIR结构域的适配蛋白诱导产生干扰素-β)相互作用负调控天然免疫反应。然而,SARM1在免疫调节方面的作用还尚不明确。虽然最初报道称巨噬细胞中产生趋化因子CCL5需要SARM1,但最近的证据表明,SARM1在巨噬细胞中不表达,且观察到的趋化因子表型是SARM1敲除的小鼠品系的背景产生的。尽管其在免疫细胞中的作用存在争议,但SARM1在轴突变性中发挥着无可争议的关键作用,并且正在成为预防或改善神经退行性疾病和创伤性脑损伤的治疗靶点。
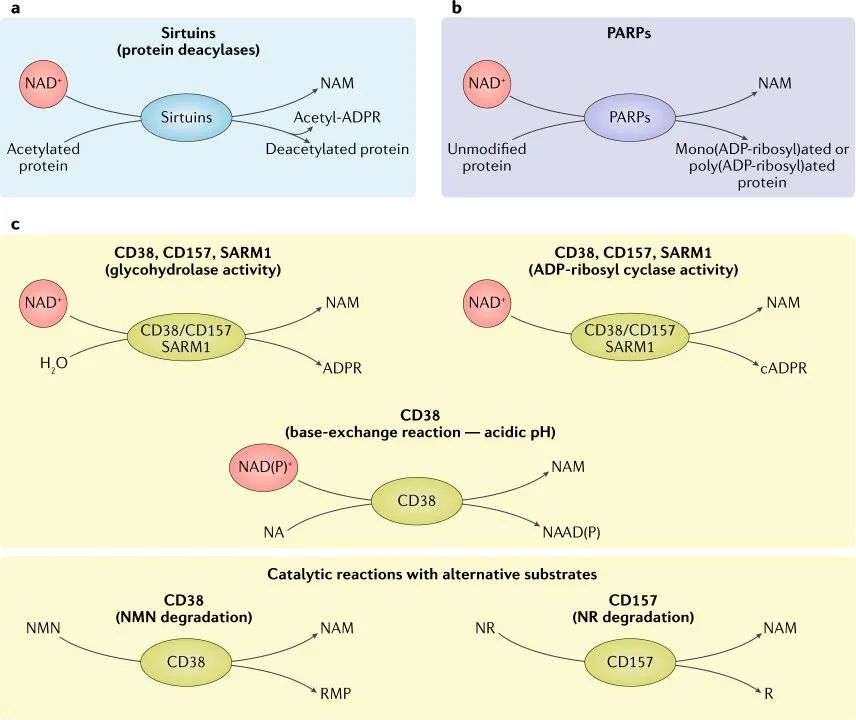
图2 | 三种主要的NAD+消耗酶。a | 乙酰化酶使用烟酰胺腺嘌呤二核苷酸(NAD+)作为辅助底物从目标蛋白上的赖氨酸残基上去除酰基。NAD+被分解,产生烟酰胺(NAM)和ADP -核糖,其中ADP -核糖作为酰基受体,产生乙酰- ADP -核糖(乙酰- ADPR)。b | 聚(ADP -核糖)聚合酶(PARP1-PARP3)使用NAD+作为单(ADP -核糖)或聚(ADP -核糖)靶蛋白的共底物,产生NAM作为副产物。c | NAD+糖水解酶与环ADP -核糖(cADPR)合成酶(CD38, CD157和SARM1)的反应。这组蛋白质的主要催化活性是将NAD+水解成NAM和ADP -核糖。在较小程度上,CD38、CD157和SARM1具有ADP-核糖基环化酶活性,从NAD+中产生NAM和cADPR。在酸性条件下,CD38也可以进行碱基交换反应,将NAD(P)+的NAM交换为烟酸(NA),生成烟酸腺嘌呤二核苷酸(磷酸盐)(NAAD(P))。据报道,CD38和CD157在其催化反应中可以使用替代底物。CD38可降解NMN为NAM和核糖单磷酸(RMP),而CD157可降解NR,生成NAM和核糖(R)。NR,烟酰胺核苷。
(3)NAD+在细胞中作用
除了作为消耗NAD+的酶相关对象外,NAD+被广泛用作生化反应的辅助因子或底物,有超过300种酶依赖NAD+发挥活性。因此,NAD+是一个关键的细胞功能和适应代谢需要的媒介。这些关键的细胞过程包括代谢途径、氧化还原稳态、维护和修复DNA以维护基因组稳定性、表观遗传调控和染色质重塑以及自噬。总的来说,这些功能对于维持系统健康和体内平衡是很重要的。然而,在衰老过程中,NAD+水平下降不仅会影响这些过程,并且会加剧衰老相关疾病发生(图3)。
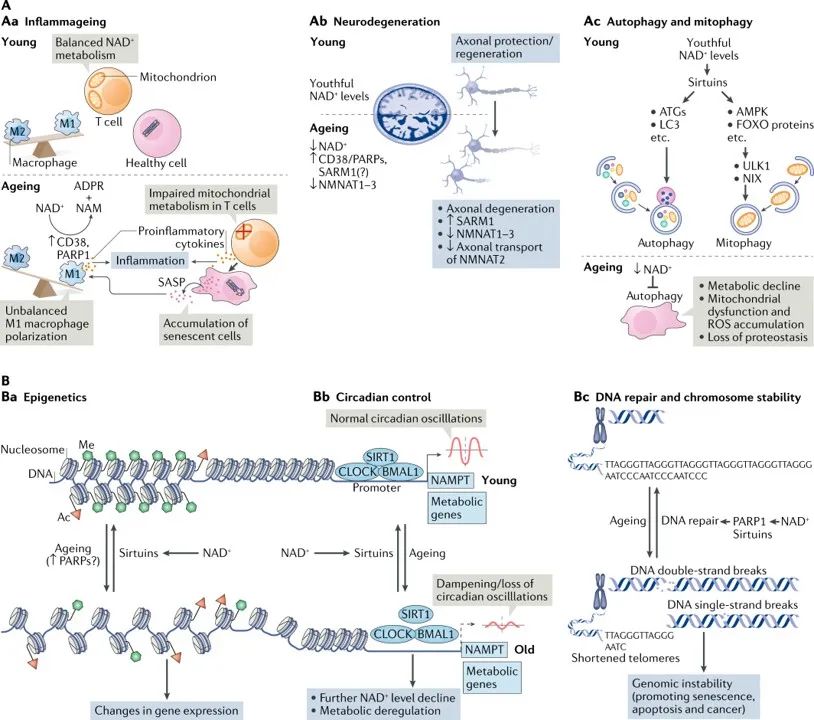
图3 | 衰老过程中NAD+代谢。A:降低烟酰胺腺嘌呤二核苷酸(NAD+)水平涉及与衰老相关的多种生物学过程。Aa :衰老与异常的促炎免疫细胞激活或“炎症”有关,导致持续的低级别炎症。这部分是由衰老细胞的积累引起的,通过衰老相关的分泌表型(SASP)促进巨噬细胞表型向促炎M1状态极化,从而导致炎症。有证据表明,在这些巨噬细胞对SASP的反应中,NAD+消耗酶CD38和多聚(ADP -核糖)聚合酶(PARPs)的表达增加,导致NAD+水平下降,这些机制对衰老过程中NAD+水平的降低起到重要作用。此外,有研究表明,在衰老的T细胞中线粒体功能下降,导致了促炎细胞因子的分泌增加,促进炎症状态,也导致衰老。Ab:轴突变性是许多年龄相关神经元疾病的前兆,其特征是快速的NAD+损耗。在正常生理条件下,NAD+生物合成酶,烟酰胺单核苷酸腺苷转移酶(NMNATs)对轴突变性具有保护作用,其表达支持轴突的维持和防止神经退行性变。特别是,NMNAT2在轴突中是一个重要的生存因子,,实现快速的周转需要不断地将它从其合成的体细胞运送到轴突,而这些运输过程在轴突变性过程中受到干扰。此外,NAD+消耗酶SARM1被轴索损伤激活,通过促进NAD+降解介导轴索变性。Ac:自噬是一个关键的细胞分解过程,允许细胞适应不同的营养可用性,并服务于细胞质量控制,允许清除缺陷细胞器和蛋白质。自噬通过Sirtuins(主要是SIRT1)调控下游的NAD+水平。NAD+水平的下降降低了整体自噬通量,以及通过线粒体自噬选择性地去除线粒体,提示自噬缺陷可能是衰老过程中NAD+消耗的结果,导致细胞功能障碍。B:因为NAD+是多种酶的辅助因子,NAD+的丢失影响多种细胞过程。例如,表观遗传调控因子如SIRT1的活性需要NAD+,其水平的下降会引起组蛋白修饰的改变,从而影响基因表达中的染色质组织和功能。有证据表明,与衰老相关的NAD+缺失与PARPs表达增加有关,这可能是由于衰老过程中DNA损伤水平的增加和DNA修复的需要(Ba)。NAD+也影响核心时钟组件clock和BMAL的转录活性,从而调节重要代谢基因的昼夜节律表达以及烟酰胺磷酸核糖转移酶(NAMPT),而这反过来是NAD+水平昼夜节律振荡所必需的(Bb)。降低的NAD+水平也干扰DNA修复中PARPs和Sirtuins的活性,导致基因组不稳定,即衰老和癌症的标志产生(Bc)。ADPR,ADP-ribose;ATG,自噬相关蛋白;FOXO,叉头盒蛋白O;ROS,活性氧。
(二)衰老中的NAD+依赖机制
在衰老过程中,NAD+水平下降,许多与NAD+降解和生物合成相关的酶发生改变。NAD+和衰老的10个特征之间的关系已被广泛回顾。此外,在衰老过程中NAD+水平的下降与衰老相关疾病的发展和进展有关,包括动脉粥样硬化、关节炎、高血压、认知能力下降、糖尿病和癌症。在本节中,作者重点讨论了影响衰老或受衰老影响的主要细胞过程,如代谢功能障碍、DNA修复失败和基因组不稳定、炎症、细胞衰老和神经退行性病变,并讨论NAD+水平对其的调控。补充NAD+可能对所有这些过程以及与之相关的衰老相关疾病都有所缓解。已有潜在的恢复NAD+水平的治疗策略,如用膳食前体恢复NAD+水平,用小分子抑制剂靶向NAD+降解酶,从而为缓解年龄相关的衰退和疾病提供了机会(图4)。
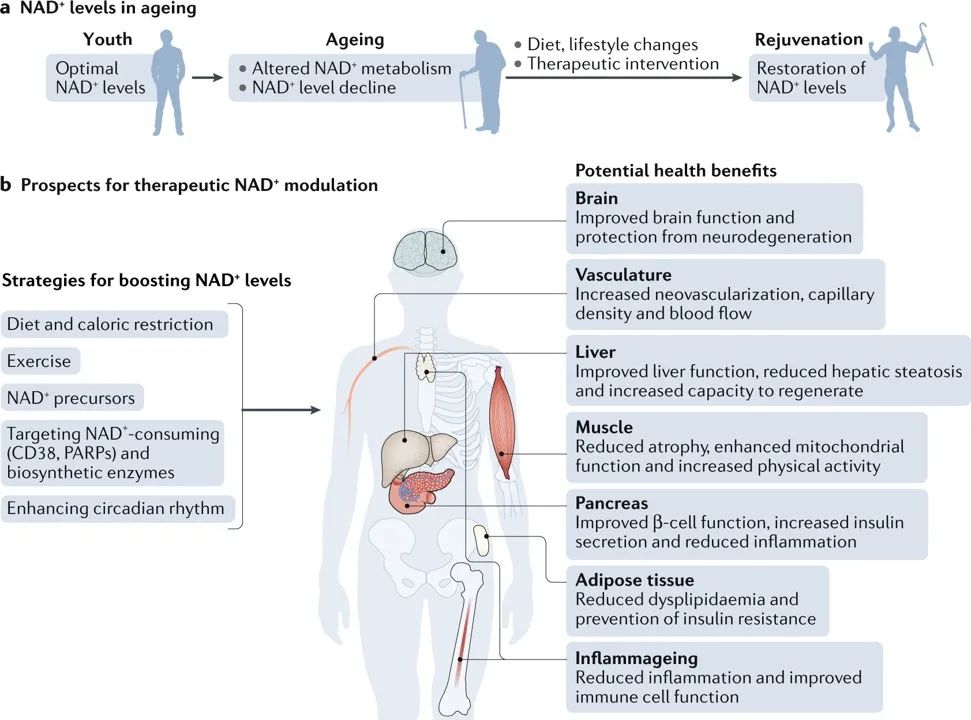
图4 | 恢复NAD+水平的治疗方法及其对健康的影响。衰老与降低烟酰胺腺嘌呤二核苷酸(NAD+)水平有关,这会促进或加剧与衰老相关的疾病。因此,恢复NAD+水平已成为预防和治疗衰老相关疾病,恢复老年人的健康和活力的一种方法 (a),目前有一些潜在的措施来提高NAD +水平,包括生活方式的改变,例如增加锻炼,减少卡路里的摄入量,每天健康饮食和遵循一个一致的昼夜节律模式、符合健康的睡眠习惯和进餐时间。另一种方法是使用小分子抑制剂或激活剂来促进NAD+的生物合成,以及使用膳食补充剂,包括NAD+前体,如烟酰胺单核苷酸和烟酰胺核糖苷。所有这些方法都能提高组织NAD+水平,对健康有益,可以改善组织和器官功能,防止认知能力下降,改善新陈代谢健康,减少炎症和增加生理益处,这些可能共同发挥作用延长病人的健康寿命和潜在的寿命(b)。
(1)代谢功能障碍
在世界范围内,肥胖是一种日益严重的流行病。肥胖的个体更有可能发展为以肥胖增加、胰岛素抵抗、高血糖水平、高血压和血脂异常为特征的代谢疾病。这些人也因此患2型糖尿病、心血管疾病、非酒精性脂肪性肝病、动脉粥样硬化、中风和癌症的风险更高。肥胖还会加速衰老,与寿命缩短有关。在过去的20年里,越来越多的证据表明靶向NAD+代谢可能提供一定的治疗作用,有助于治疗代谢性疾病及延缓衰老。
NAD+最初是通过调节酵母提取物的代谢速率而被发现的,至今NAD+与代谢之间的联系已经被发现了近一个世纪。作者现在知道,NAD+处于代谢的核心,并调节多种代谢途径的代谢通量(详见框1)。因此,NAD+稳态对于各种代谢组织的正常功能是必需的,包括脂肪、肌肉、肠道、肾脏和肝脏。然而,最近在酿酒酵母中另一个关键发现表明,长寿蛋白Sir2(酵母去乙酰化酶)以NAD+依赖的方式延长了寿命,这表明Sir2活性可能与代谢状态有关。通过对哺乳动物系统寿命延长代谢操作,如运动、卡路里限制、限时饲养和生酮饮食,和健康的生理规律一样 (包括常规睡眠模式)都可以增加一部分NAD+工作水平,从而导致去乙酰化酶(Sirtuins)的激活,这支持了上述的假说。
而改变或破坏代谢状态,例如高脂饮食、产后体重丧失和昼夜节律的破坏都可导致NAD+水平降低,从而降低去乙酰化酶(Sirtuins)活性以及其他依赖NAD+的细胞过程。相反,增加细胞NAD+水平最近被证明可以减少压力并驱动代谢反应的方向。此外,较高的NAD+水平可以促进核SIRT1和线粒体SIRT3的去乙酰化酶活性,从而调节线粒体功能,保护机体免受高脂饮食诱导的代谢性疾病的侵袭。
综上所述,这些关键研究提供了强有力的证据和理论基础,即靶向NAD+降解途径或提高NAD+水平可影响代谢过程,并可预防代谢疾病。目前已有大量研究进一步支持这一模型,研究发现Parp1敲除和Cd38敲除小鼠或经PARP/CD38抑制剂处理的小鼠具有超生理水平的NAD+,在高脂饮食条件下和在衰老过程中也不会发生肥胖,且有较高的代谢率和(相对)正常的葡萄糖代谢。此外,接受高脂饮食的小鼠炎症增加,导致NAMPT表达降低,而NAD+逆转了通路活性的降低 (框1),为肥胖期间NAD+水平下降提供了一个潜在的机制解释。脂肪细胞特异性缺失NAMPT的小鼠脂肪组织中NAD+水平降低,胰岛素抵抗增加,代谢功能紊乱加剧,可以通过补充NMN来恢复。此外,近期多项研究表明,NAD+补充剂(NR和NMN)可以恢复与衰老相关的低NAD+水平,在啮齿类动物模型中也可以防止肥胖,这提示这些补充剂可以作为治疗手段来恢复人类肥胖患者的代谢健康。最近的几项临床试验已经开始研究NAD+前体在人类肥胖患者改善代谢健康和糖代谢方面的疗效。到目前为止,这些研究大多是在健康人群中进行的(见表1),以测试NR和NMN的有效剂量是否可以安全地提高NAD+水平。然而,最近的两项随机和双盲研究用NR治疗超重和肥胖患者分别为6周和12周。不幸的是,虽然NR可以有效地增加这些个体的NAD+水平,但两项研究的参与者都没有任何体重减轻、胰岛素敏感性增加或线粒体功能增强的迹象。因此,靶向NAD+代谢是否能有效治疗肥胖或老年人的代谢性疾病还不清楚。
(2)免疫细胞功能异常
炎症现在被认为是衰老的标志和疾病的关键驱动因素(图3Aa)。慢性炎症通过免疫细胞和代谢细胞(如肝细胞和脂肪细胞)之间复杂的相互作用对全身代谢产生深远影响,如葡萄糖和脂质摄取以及胰岛素敏感性。尽管在过去的十年中,人们对免疫代谢越来越关注,但对于NAD+如何影响慢性炎症和免疫细胞功能却知之甚少。
先天免疫
慢性低度炎症现在被认为是衰老相关疾病和代谢疾病的关键驱动因素,表现为先天免疫系统异常激活,促炎细胞因子表达增加,如肿瘤坏死因子(TNF)、IL-6和IL-1β,和激活免疫复合物,如NLRP3炎症小体。此外,现在发现巨噬细胞激活状态的改变和表型极化是这种炎症的关键来源。例如,在肥胖组织(如内脏脂肪)中,应激脂肪细胞招募了外周单核细胞,导致促炎M1样巨噬细胞进行性浸润,并取代了抗炎M2型巨噬细胞。内脏脂肪中巨噬细胞极化状态的这种转变伴随着促炎细胞因子表达增加胰岛素抵抗和脂解率降低。巨噬细胞的发现者Elie Metchnikoff在100多年前首次在衰老组织中观察到巨噬细胞数量的增加。尽管这一现象最初被忽视,但现在越来越多的证据表明,衰老不仅导致巨噬细胞数量增加,而且伴随着巨噬细胞极化状态和功能的改变,这是炎症的关键驱动因素。一些最早提出NAD+影响巨噬细胞功能的研究表明,抑制NAMPT和随后巨噬细胞中NAD+池的消耗均减少了促炎细胞因子(如TNF)的分泌,并导致巨噬细胞形态学改变。最近的研究表明,NAD+是巨噬细胞功能的关键调节因子,巨噬细胞的激活与NAD+生物合成或降解途径的上调有关。例如,本文作者团队最近证实促炎(M1)巨噬细胞极化与CD38表达增强相关,导致NAD+消耗增加。相反,抗炎(M2)巨噬细胞极化与依赖于NAMPT45的NAD+水平增加相关。在M1和M2巨噬细胞中阻断NAM挽救通路(表1)均显著降低了与M1和M2表型相关的选择基因的表达。NAD+的前体NMN和NR可以绕过和挽救NAMPT的抑制,从而挽救这一效应。M2巨噬细胞比M1巨噬细胞需要更多的NR/NMN来挽救巨噬细胞的活化,说明NAD+是一般巨噬细胞活化的关键代谢产物,其代谢在M1和M2巨噬细胞中受到差异调控,控制不同的生物学过程和功能。与作者的研究结果一致的是,最近的一项研究发现,M1巨噬细胞极化与NAD+的降解增强有关,抑制NAMPT可阻断M1巨噬细胞的糖酵解转移,限制体外促炎反应,减少脓毒症反应中的体内全身炎症。本研究和最近的另一份报告表明,这种NAD+的转换,特别是在巨噬细胞M1极化的最初几个小时,依赖于活性氧诱导的DNA损伤和PARP1的激活。然而,作者实验室最近发表的结果没有发现在M1巨噬细胞极化过程中DNA损伤或PARP1激活的证据。
相比之下,CD38是M1巨噬细胞中主要的NAD+消耗酶。这些结果与最近的研究结果一致,表明M1巨噬细胞由于参与抗氧化防御的基因的转录增加,如SOD2,可以免受活性氧诱导的DNA损伤。因此,这些矛盾的现象说明需要更好地描绘在促炎和抗炎巨噬细胞极化时NAD+的消耗,明确观察到的NAD+水平的下降及其发生环境和时间,以及NAD+基因表达水平是否影响促炎和抗炎巨噬细胞功能的分子机制。
在衰老过程中,NAD+水平下降与肝脏和脂肪中促炎M1样巨噬细胞的增加积累有关,其特征是CD38表达增加和NADase活性升高。通过体外和体内方法,作者发现这些过表达CD38的M1样巨噬细胞被衰老细胞分泌的炎性细胞因子直接激活。此外,衰老的巨噬细胞的特征是NAD+的新生合成受损,而NAD+本身可能会影响衰老过程中巨噬细胞的功能。
在炎症方面,上述研究提示促炎M1样巨噬细胞可能是衰老组织中促炎细胞因子的主要来源。衰老与NLRP3炎性细胞的激活增加相关,该炎症细胞主要由髓系免疫细胞表达,随后IL-1β的表达增加。促炎细胞因子的表达增强可能会推动炎症的恶性循环,导致更严重的炎症,增强组织和DNA损伤,进一步激活NAD+的消耗,如CD38和PARPs,并加速与年龄相关的生理衰退。因此,靶向巨噬细胞免疫代谢通路,特别是调控NAD+生物合成通路或降解通路,可作为激活或抑制巨噬细胞功能、调节巨噬细胞极化状态的治疗策略。这当然是和调节免疫衰老相关的,但也可以用来缓解由慢性炎症疾病,神经退行性疾病和自发炎症疾病,也可作为癌症治疗策略。
适应性免疫
像先天免疫系统一样,衰老的特征是由于适应性免疫细胞功能的改变而导致建立有效适应性免疫应答的能力降低,这一过程被称为免疫衰老。
衰老会导致免疫细胞群的失衡,包括幼稚T和B细胞水平降低,T细胞抗原受体多样性丧失以及虚拟记忆T细胞(virtual memory T cells)水平升高。NAD+和NAD+消耗酶在T细胞生物学中的调节作用已经被证明了;然而,它们对适应性免疫衰老的贡献在很大程度上是未知的。一方面,细胞外NAD+被认为是引起特定T细胞亚群如调节性T细胞中细胞死亡的危险信号。另一方面,NAD+似乎表现出免疫调节特性,例如影响T细胞极化。但是,NAD+是否能促进特定的T细胞表型以及NAD+前体对NAD+代谢的操纵是否会导致类似的免疫调节特性仍是未知的。
适应性免疫衰老的既定标志是具有高度细胞毒性的CD8+ CD28-记忆T细胞群的扩展,以效应分子(例如颗粒酶B)的高分泌为主要特征,区别于其他细胞群。这种细胞群的特征是SIRT1和FOXO1水平的降低、糖酵解能力增强和颗粒酶B产生增加。这些研究强调了在年龄相关的适应性免疫功能障碍中通过操纵NAD+相关途径进行代谢重编程的潜力。通过抑制CD38,使NAD+-SIRT1-FOXO1轴上调从而增强了Th1/Th17杂交细胞中的效应子功能,未来使用CD38抑制剂或NAD+前体的研究可能揭示在衰老的适应性免疫系统中靶向NAD+的治疗潜力。
适应性免疫衰老的另一个免疫学特征是耗竭的T细胞数量的增加,其特征是抑制性受体分子(例如PD1和TIM3)的表达,增殖能力降低和效应子功能降低。PD1是免疫检查点的组成部分,通常将阻断PD1作为一种抗癌策略,但也有人提出可以恢复老年T细胞的效应子功能。关于NAD+代谢,最近的一项研究表明,在PD1阻滞性癌症中,CD38过表达和功能失调与CD8+T细胞耗竭有关,这突出了将CD38抑制研究扩展至与年龄相关的T细胞耗竭的潜力。然而,该假设尚待深入探讨,需要更多的研究来确定这种方法的临床前疗效。
因此,总的来说,需要做更多的工作来确定操纵NAD+水平在逆转适应性免疫系统中与衰老相关的免疫功能障碍方面是否有效,同时明确这种操纵是否安全同等重要。
(3)细胞衰老
在衰老期间,暴露于代谢、基因毒性或癌基因诱导的应激的细胞会经历不可逆的细胞周期停滞,称为细胞衰老。衰老细胞的一种主要表型是炎症介质(主要是细胞因子和趋化因子)的表达增加,称为衰老相关的分泌表型(senescence-associated secretory phenotype,SASP),这也被认为是一种促进疾病产生的方式。它通过干扰干细胞更新、组织和伤口的修复和促进炎症产生,从而导致组织稳态的降低(图3Aa)。随着年龄增长,衰老细胞的数量逐渐增加,细胞衰老与多种和年龄相关的疾病有关,而药理学上的镇静剂清除衰老细胞可能是一种有效的治疗方法,可治疗多种以前无法治愈的疾病,包括阿尔茨海默病。此外,在衰老过程中提高细胞NAD+水平的治疗方法有望延长健康期,但尚不清楚NAD+如何影响细胞衰老。最近研究显示,衰老细胞的NAMPT表达上调(表1),以及SASP均取决于NAD+水平。用NMN处理衰老细胞可以增强SASP,导致慢性炎症增加,并且促进炎症驱动的癌症的发展。这些研究结果表明,NAR+增强剂(如NR和NMN)的给药可能以长期副作用为代价,例如促进慢性炎症和癌症的发展。因此,更多地了解提高NAD+水平的益处和其他副作用将是未来研究和正在进行的临床试验中重点关注的领域。由于炎症是一个非常复杂且多效的过程,因此需要进一步研究以更好地了解NAD+水平在怎样的条件下如何影响不同的炎症状态,并确定NAD+代谢以怎样的机制影响炎症性免疫和衰老细胞的生物学问题。
尽管有很多文献记载了在衰老组织中观察到衰老细胞的积累,并且这些组织中伴随着NAD+水平的下降,但尚无研究将衰老过程中炎症性衰老细胞的积累与NAD+水平联系起来。最近,有研究表明,随着年龄的增长,哺乳动物组织中CD38含量增加,并且已经提出CD38是导致NAD+含量下降的主要NAD+消耗酶。但是, 在衰老组织中驱动CD38表达增加的机制以及在这些组织中表达CD38的细胞类型尚不清楚。最近的观察表明,先天免疫细胞,特别是巨噬细胞,可能是对SASP产生NAD+降解的主要细胞群,从而导致了整个生物体NAD+水平的下降。作者的研究表明,在衰老细胞中共培养或在衰老细胞的条件培养基中培养的巨噬细胞出现了NAD+消耗性酶CD38的表达增强和增殖增加。重要的是,另一小组也独立地表明衰老细胞及其SASP激活巨噬细胞中CD38的表达并促进CD38依赖性NADase活性。此外,在衰老的小鼠模型和经衰老诱导化疗剂阿霉素治疗的小鼠中发现,内脏脂肪和肝脏等新陈代谢组织中积累的衰老细胞可以直接激活组织常驻巨噬细胞中CD38的表达。为了进一步支持炎症、衰老细胞负担和NAD+之间的联系,最近对小鼠的研究还表明,线粒体功能异常的细胞会启动促炎程序,并分泌促炎细胞因子,这与更大的衰老细胞负担、新陈代谢和身体机能障碍以及过早衰老有关。在这种情况下,补充NR能够通过减轻炎症和衰老细胞负担来部分挽救这种多发病综合征,这与上述NAD+前体可以增加SASP表达的发现相反。因此,NAD+对衰老的调节似乎很复杂。
总体而言,这些发现表明,在旨在恢复衰老过程中NAD+水平的方法中,应考虑以免疫细胞(例如T细胞、巨噬细胞和衰老细胞)作为靶标。但是,在对增强NAD+水平的长期副作用有更多了解之前,这项工作应谨慎进行开展。
(4)神经变性
衰老与大多数神经退行性疾病密切相关,并伴随着哺乳动物大脑中细胞NAD+水平的降低。在加速衰老的几种模型中都报告了NAD+消耗的现象,这些模型表现出神经退行性变,以及神经退行性疾病,包括阿尔茨海默病、帕金森病和肌萎缩性侧索硬化(ALS)。在与年龄有关的神经退行性疾病期间,大脑中NAD+损失的根本原因和机制仍然未知。但是,有许多证据支持NAD+具有神经保护作用(图3Ab)。
首先,轴突变性是许多与年龄有关的神经元疾病的先兆,其特征是NAD+的快速耗竭。在正常生理条件下,NAD+生物合成酶烟酰胺单核苷酸腺苷酸转移酶2(nicotinamide mononucleotide adenylyltransferase 2,NMNAT2)(图1;表 1)是轴突的生存因子,由于其快速转折,需要通过顺行轴突运输对其进行持续补充。但是,在轴突变性过程中,NMNAT2轴突运输受阻,轴突中的蛋白质池迅速降解,导致轴突中NAD+的严重消耗。而且,消耗NAD+的酶SARM1被轴突损伤激活并通过促进NAD+降解介导轴突变性。这最初显示在Wallerian慢变性(Wallerian degeneration slow,Wlds)小鼠中,该小鼠受到轴突变性的保护,显示出SARM1表达的缺失和较高的神经元NAD+水平,这是由于NMNAT1嵌合融合蛋白的过表达,它可以从细胞核重新分配到轴突,在那里它可以替代NMNAT2的活性。SARM1在促进轴突损伤中的作用已在其他几种体内模型中得到证实。Sarm1基因敲除小鼠能够免受轴突变性的侵害,且可以挽救由于缺乏NMNAT2引起的严重轴突生长缺陷和围产期死亡。最近,在神经元中过表达显性负性表达的SARM1转基因小鼠模型显著延迟了轴突变性,提示基因治疗或靶向SARM1的小分子有望治疗神经疾病。
总体而言,Wlds小鼠研究在很大程度上显示了生物合成酶NMNAT1、NMNAT2和NMNAT3的神经保护功能,以及它们在包括帕金森病在内的几种神经退行性疾病中的保护作用。但是,机制尚不清楚。除了上面讨论的SARM1依赖性NAD+降解调控外,NMNAT降解自身底物和NAD+前体NMN似乎还可以保护轴突免于变性。与NAD+的神经保护作用相反,据报道前体NMN具有促进SARM1活化并环化ADP-核糖,导致轴突破坏的神经毒性作用。但是,NMN积累导致轴突变性的证据需要验证,并且仍存在争议,但这种神经毒性作用的可能性还是引发了对通过补充NAD+前体(例如NMN)来增加NAD+合成的治疗潜力的质疑。
支持NAD+具有神经保护作用的其他证据还包括使用P7C3的研究,P7C3是一种氨基丙基咔唑,据报道是NAM挽救途径中NAMPT的变构激活剂(框1)。在帕金森病、阿尔茨海默病和ALS158的小鼠模型中,P7C3被证明具有神经保护作用。此外,据报道,除SARM1外的NAD+消耗酶在衰老相关的神经退行性疾病中发挥消耗细胞内NAD+的作用。例如,CD38的表达在阿尔茨海默病进展过程中增加,而缺乏CD38的阿尔茨海默氏病小鼠模型(Cd38-knockout mice)大脑中NAD+水平升高,表现出较轻的疾病表型。与NAD+的神经保护作用一致,Cd38敲除小鼠也受到保护,免受缺血性脑损伤后神经元死亡的影响。脑中的多种细胞类型,包括小胶质细胞、星形胶质细胞、神经元和内皮细胞均表达CD38,用炎症细胞因子处理小胶质细胞和星形胶质细胞会诱导CD38的表达。与这一发现一致的是,CD38的表达与小鼠大脑中伴随着大量炎症性巨噬细胞/小胶质细胞的神经炎症有关。据报道,CD38及其同系物CD157也会影响社会行为,这证实了NAD+对神经元功能的功能影响。尽管没有直接证据表明CD38在神经退行性疾病中具有因果作用,但如上所述,CD38逐渐成为与炎症和衰老密切相关的关键酶,且与神经退行性疾病密切相关。最后,PARP1的激活还与阿尔茨海默病和帕金森病的发病有关。使用各种阿尔茨海默病和帕金森病模型的体内研究表明,PARP1的缺失可防止脑功能障碍和认知功能下降。然而,在神经退行性变过程中PARP1激活对NAD+耗竭的贡献尚待探索。
综上,越来越多的证据表明NAD+是维持健康神经系统的主要代谢物,并且可以影响多种脑细胞类型的生物学,这表明抵消与衰老相关的NAD+水平下降可能是一种可行的治疗方法。研究发现使用NAD+补充剂恢复NAD+水平以及两种生物合成酶NAMPT和NMNAT1的过表达可防止轴突变性。此外,NAD+前体NR和NMN改善了阿尔茨海默病大鼠和小鼠模型中的神经元细胞健康、记忆和认知功能,并在帕金森病的果蝇黑色素体模型和ALS139小鼠模型中显示了神经保护特性。重要的是,目前几项使用NAD+前体(尤其是NR)的临床试验正在进行中,以期用于治疗神经系统疾病和促进健康衰老(表 2),这些试验无疑将拓展人们对人类神经退行性过程中NAD+代谢的了解。
(三)提高NAD+水平的目标及策略
在过去的二十年中,NAD+在健康衰老和长寿中的重要性已得到认可。在不同动物模型(如秀丽隐杆线虫、黑线虫、啮齿动物和人类原代细胞)中进行的临床前研究已明确表示,NAD+含量随年龄而下降,其下降幅度为10%至65%,具体取决于不同的器官和年龄。 NAD+可以通过饮食和生活方式选择来调节(图4),也可以通过药物调节,到目前为止,已经探索出了三种增加NAD+水平的主要方法:从饮食途径补充参与NAD+挽救通路的NAD+前体; NAD+生物合成酶的调节,特别是那些调节从头合成和挽救途径的限速步骤的酶(分别为α-氨基-β-羧基粘康酸酯ε-半醛脱羧酶(α-amino-β-carboxymuconate ε-semialdehyde decarboxylase ,ACMSD)和NAMPT); 以及与NAD+降解有关的酶(如PARPs和CD38)的抑制作用。 NAD+水平的提高已在多种人类疾病小鼠模型(表 1)中显示出功效,从而导致了衰老期间人类对NAD+增强剂的众多临床试验(表 2)。
啮齿动物的大多数临床前研究表明,NAD+增强疗法具有很强的临床转化潜力。但对人体的研究尚不成熟,迄今为止,评估NAD+前体的药代动力学和毒理学的临床试验初步表明,NMN和NR的给药是安全的,可以有效提高健康志愿者中NAD+的水平(表 2)。值得注意的是,使用NR进行的I期临床试验比使用NMN进行的临床试验更多,这些试验结果相互矛盾。一项试验表明,短期NR给药对健康的老年人有一定的有益作用,另一项试验对ALS患者也显示出积极的结果。但是,NR对肥胖的老年男性几乎没有影响(另请参见“新陈代谢功能障碍”和“表 2”部分的讨论)。因此,需要进一步的人类临床研究以确定合适的剂量,治疗时间和长期的毒理学结果,并考虑参与者的多样性,以更好地助推NAD+增强策略的转化。
(1)膳食补充
不同NAD+前体对酵母和秀丽隐杆线虫的寿命和健康的影响已经得到了广泛研究。在野生型酵母和秀丽隐杆线虫中,低(微摩尔)浓度的NR以去乙酰化酶依赖性方式延长寿命。另外,在野生型秀丽隐杆线虫上施用NAM 观察到寿命延长。但是,NAM的超生理剂量(1-5 mM,而在延长寿命的实验中使用的200 μM)也与酵母和秀丽隐杆线虫的寿命缩短有关。因此,不清楚NAM在衰老过程中是有益还是有害的。这种差异的一种可能解释是NAM可以直接和间接地影响NAD+功能。 除了作为挽救途径中的直接NAD+前体,NAM还是NAD+分解代谢的副产物,并且在高浓度下,可作为NAD+依赖酶(如 PARPs和去乙酰化酶)的反馈抑制剂。但是,有关高剂量NAM抑制作用的结果在一些动物研究中尚无定论。NAM抑制NAD+活性的另一个可能原因可能是NAM水平升高导致NAM甲基化成比例增加。反过来,这会影响甲基的细胞利用率,这对于DNA甲基化和基因表达调节(框2)很重要。高水平的甲基化NAM与2型糖尿病、帕金森氏病和心脏病的发病机制有关。因此,需要进一步的研究来阐明最安全的NAM剂量和治疗时间。
增强NAD+的策略也已被用于延长患年龄相关疾病患者的寿命。使用NR和/或NMN的NAD+补给还有效延缓了A型干性皮肤色素性干燥、共济失调性毛细血管扩张和Cockayne综合征的相关秀丽隐杆线虫模型中的早衰表型,并延长了以上模型和一种Werner综合征的蠕虫模型的寿命。NMN治疗在共济失调毛细血管扩张症小鼠模型中也显着提高了NAD+水平和最大寿命。在肌肉营养不良的小鼠模型中,NR的治疗还能通过调节线粒体代谢来改善肌肉干细胞的功能。
迄今为止,很少有研究调查NMN、NAM和NR对延长野生型小鼠的健康寿命和/或寿命的影响。在一项研究中,从5个月大开始长期(1年)喂养NMN小鼠可提高全身胰岛素敏感性,能量代谢和体力活动,并改善血脂状况,而没有明显的毒性作用。在另一项研究中,长期施用NAM可以预防与高脂饮食有关的疾病,促进健康,但对寿命没有显著影响。最后,在老年小鼠中服用NR可以延长寿命(5%),但幅度很小。这些结果反映出大多数使用NAD+前体的临床前研究都集中于通过抵消以NAD+水平下降为特征的各种与年龄相关的疾病(表 1)。研究表明,NMN增强了各种器官的线粒体功能,包括骨骼肌、肾脏、肝、心脏、眼睛、脑和心血管系统,并且可以减轻血管的氧化应激,包括老年大脑皮层的神经血管耦合反应的改善。NMN补充的这些作用可能是由去乙酰化酶介导的。例如,SIRT3介导NMN诱导在心肌病小鼠模型中的心脏和心外代谢功能改善。补充NMN还可以通过依赖SIRT1的机制增加老年小鼠的血流量和耐力,从而增加内皮细胞的数量并改善内皮细胞的功能。
最后,已经表明,还原形式的NR、NRH,比NR在增加细胞内NAD+含量方面更稳定和有效,这是通过新的代谢途径发生的,其中腺苷激酶将NRH转化为NMNH,然后被氧化为NNN 或进一步被NMNAT代谢产生NADH,然后产生NAD+。因此,需要进一步的研究来比较相对于更常用的NAD+前体NAM,NR和NMN使用NRH作为治疗药物的功效,以及其潜在的副作用。
总之,以上讨论的有关NAD+前体的临床前结果表明,NAD+的增强可以改善哺乳动物的健康状况甚至延长寿命。NAD+前体在其它动物中表现出的这些有益作用可能在人类身上也有用,目前正在临床试验中进行测试(表 2)。
(2)NAD+生物合成的调控
NAD+的挽救和从头合成途径也是提高体内NAD+水平的疗法的潜在靶标。 具体而言,已提出了NAMPT活化剂,NAD+清除途径中的限速酶和NMNATs(它们参与于NAD+从头合成和挽救途径)(图1),被认为是提高组织NAD+水平的可能的治疗手段。 实际上,神经保护剂P7C3增强了经阿霉素处理的人细胞中的NAMPT活性并提高了NAD+水平,表明它可能是衰老和与年龄有关的疾病过程(如神经退行性疾病)的功能性治疗剂。 但是,P7C3在增加NAMPT活性方面的真正功效仍然存在争议。最近,有人提出了另一种小分子SBI-797812作为有效的NAMPT活化剂,其活性在纳摩尔范围内。这种化合物增加了NAMPT介导的NMN的体外生产,重要的是增加了细胞系和体内的NAD+水平。 尽管SBI-797812在体内仅提高肝脏NAD+水平的作用有限,但这是提高细胞内NAD+水平的有前途的药理方法。 此外,TES-991和TES-102524对ACMSD的药理抑制作用增强了从头开始的NAD+合成和SIRT1活性,最终增强了小鼠肝、肾和脑中线粒体的功能。 尽管这些策略有望提高NAD+的治疗水平,但仍然存在的挑战之一是,更精确地确定其中一些分子(例如P7C3)如何在分子水平上发挥作用,确定其靶标并探索潜在的脱靶效应。
除了调节NAD+水平外,近年来,人们还主要通过施用氧化还原循环醌β-拉帕酮(一种NAD(P)H:醌的外源底物)来调节NAD+ / NADH氧化还原平衡。 受体氧化还原酶1(NQO1),可从NADH再生NAD+。NQO1被显示为受热量限制(虽然不足以介导这种饮食干预的抗衰老作用),并且通过施用β-拉帕酮来增强NQO1的活性,改善线粒体功能障碍,可以防止老年小鼠运动和认知功能下降。然而,NQO1通常在大多数实体瘤中过表达,在这些实体瘤中,β-拉帕酮给药会导致氧化还原循环和氧化应激的失衡。 因此,β-拉帕酮视情况而定可能有多种作用,应谨慎服用。
(3)抑制NAD+消耗
靶向NAD+降解酶和途径可能是最近治疗学发展中最引人注目的领域。 特别是,针对PARPs和NADase,包括CD38、CD157和SARMs,具有治疗与NAD+水平下降相关的年龄相关疾病的巨大潜力。 在高糖条件下的秀丽隐杆线虫共济失调毛细血管扩张症模型中,PARP抑制可导致野生型蠕虫的寿命显著延长。
PARP1抑制剂(例如Olaparib和Rucaparib)已作为癌症的化学疗法或单药治疗的辅助产品上市。 它们使肿瘤对DNA损伤敏感,但由于毒性而用途有限。 CD38是哺乳动物中主要的NADase之一,并且在与年龄相关的NAD+水平下降中起关键作用(请参见“ 衰老中的NAD+依赖性机制”一节)。 CD38的几种抑制剂已经存在或正在开发中,其中一些会增强体内NAD+的水平。 例如,芹菜素是一种天然存在的类黄酮,可增强人肥胖症小鼠模型中人细胞和小鼠肝脏组织中NAD+的水平,并改善葡萄糖水平和脂质体内稳态。 最近,芹菜素已被证明可以下调CD38的表达,并增加糖尿病大鼠肾脏中细胞内NAD+ / NADH的比例以及SIRT3介导的线粒体抗氧化酶的活性。 另一种黄酮类CD38抑制剂木犀草苷可提高小鼠心肌缺血后的NAD+水平并保护内皮和心肌。 尽管在这种情况下未直接评估NAD+水平,临床上发现木犀草素对自闭症儿童具有神经保护作用。 另外,4-氨基喹啉的几种衍生物,包括化合物78c,抑制小鼠肌肉、肝脏和心脏中的CD38并提高NAD+水平。 化合物78c还可以防止小鼠中与年龄相关的NAD+水平下降,并且用该化合物治疗衰老小鼠可改善代谢功能障碍,减少DNA损伤积累并改善肌肉功能。 尽管该研究的作者没有直接阐述寿命变化,但他们确实发现了78c可以促进长寿期间AMPK途径的激活并降低mTOR–p70S6K和ERK通路的激活水平。 最近发现,化合物78c保护小鼠免受缺血后内皮和心肌细胞受损的伤害。 总体而言,越来越多的证据表明,靶向CD38和相关的消耗NAD+的酶(如PARPs)具有通过增强NAD+的靶标来延长人类健康寿命的潜力,并且几种抑制CD38及其NADase活性的药理方法正在开发之中( 表1)。
表1:使用NAD+增强策略的人类疾病小鼠模型的临床前研究

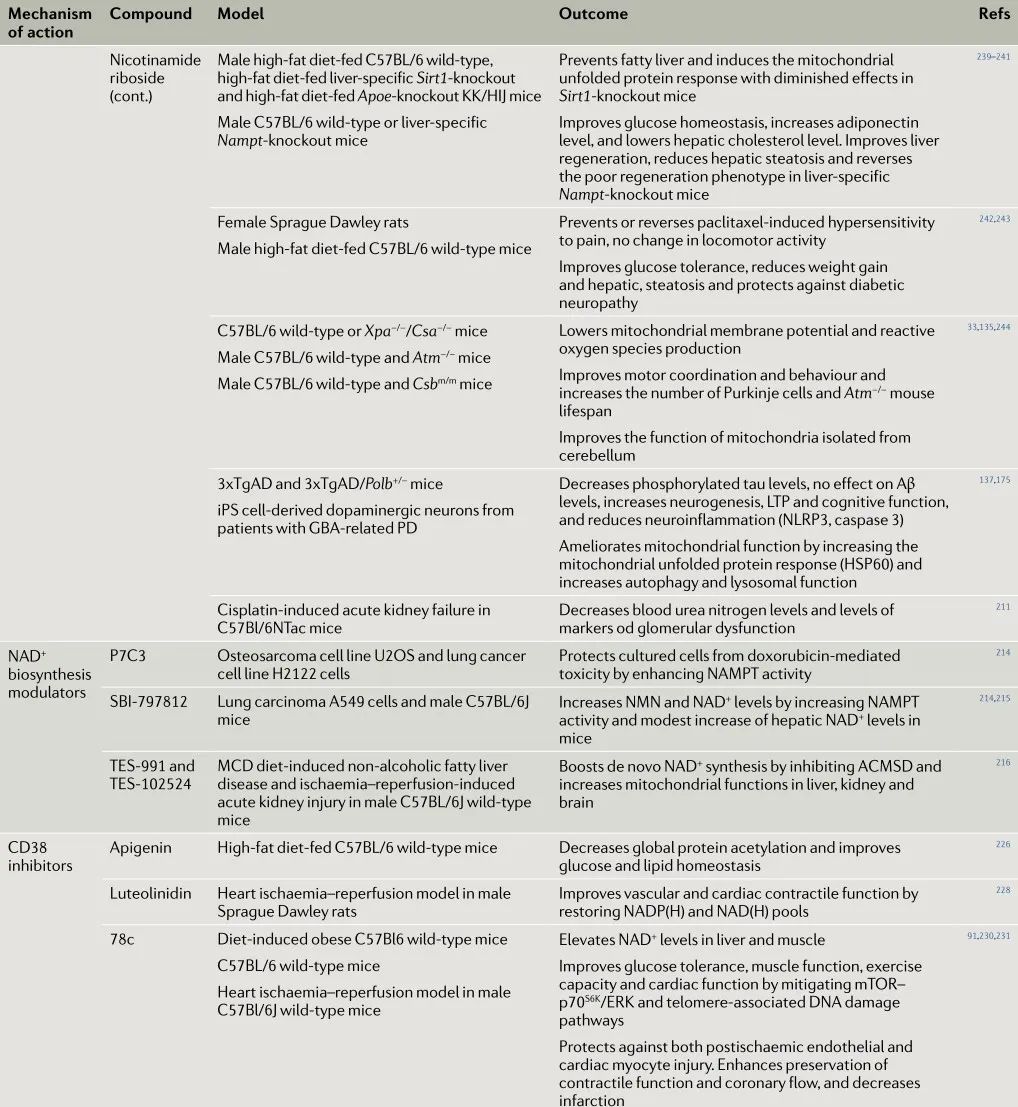
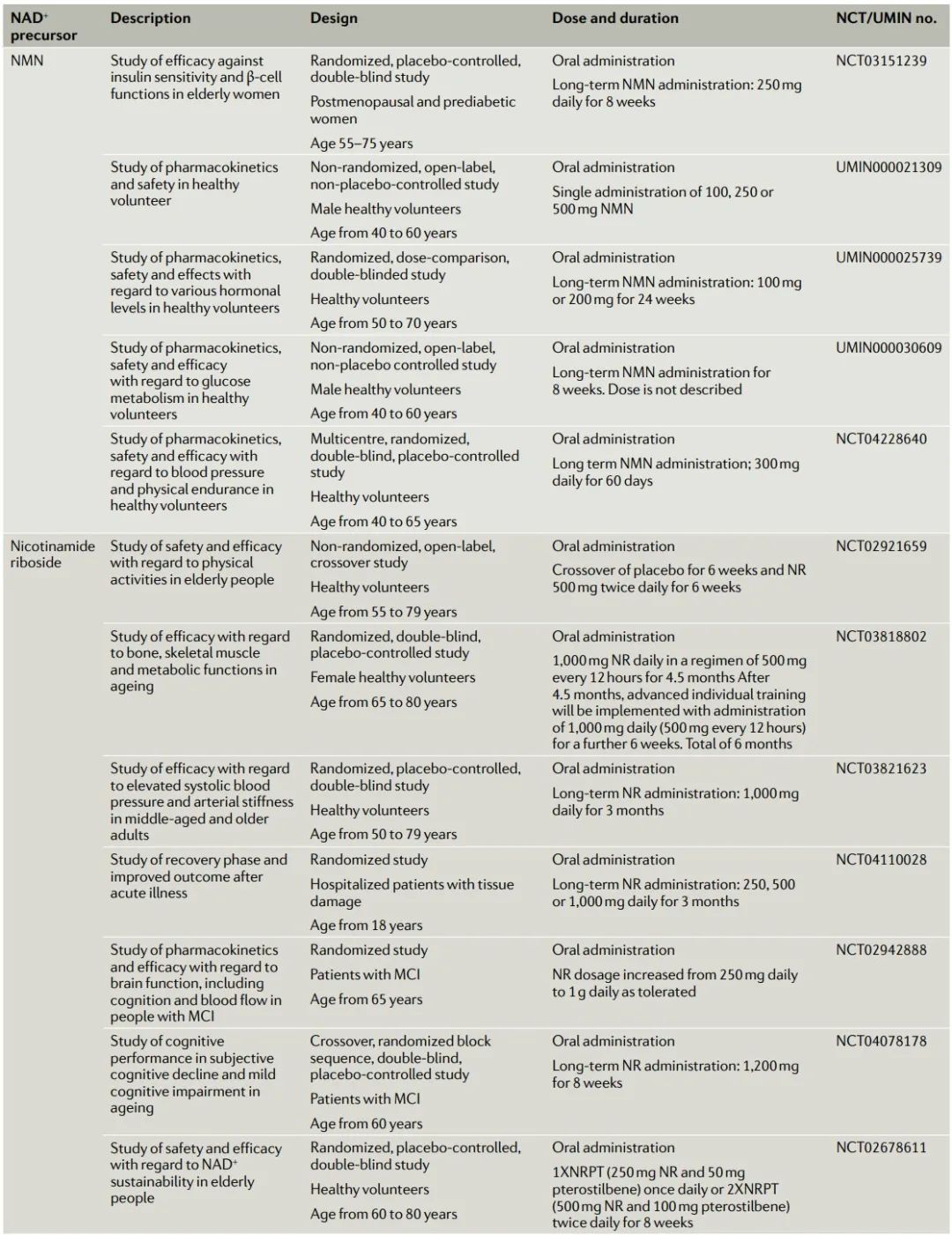
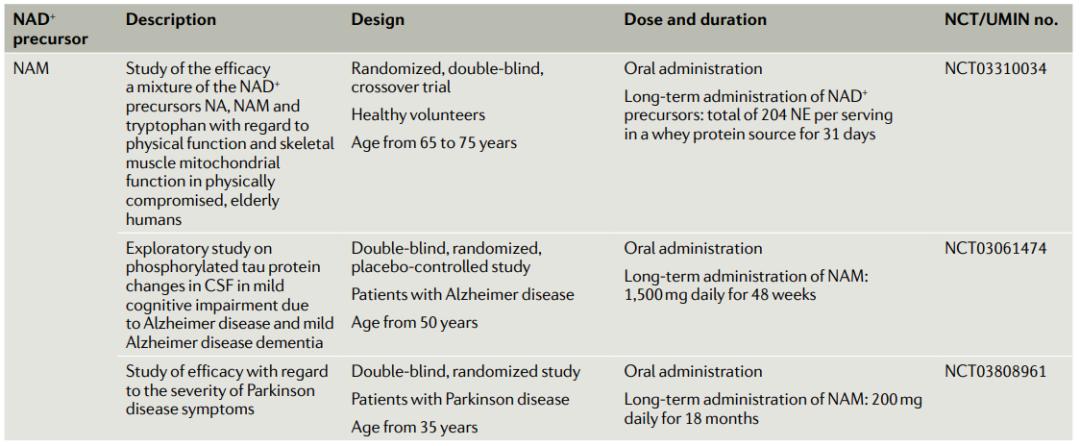
表2:关注衰老的人体临床试验
(四)总结与展望
从最初发现后的将近90年间,NAD+已成为衰老领域研究的一种重要代谢产物,并且NAD+水平下降已成为多种与年龄有关的疾病的既定特征。NAD+领域发展迅速,已发展成为生物医学研究中令人兴奋的主要研究领域之一。 在过去的5年中,该领域的研究已经有了许多进展,其中包括创新高效的工具和技术的发展,这些进一步加深了人们对NAD+水平如何影响或受复杂的信号传导、代谢和细胞通路影响的理解。 此外,通过使用稳定同位素示踪和NAD+生物传感器,人们对在细胞水平和系统水平上如何调节NAD+水平也加深了理解。 现在,人们对导致NAD+水平随年龄下降的机制以及消耗NAD+的酶(例如CD38和SARM1)以及PARPs在此过程中的新兴作用有了更深入的了解。 此外,现在已知NAD+水平的这种下降会影响多种年龄相关的细胞过程,包括DNA修复、氧化应激和免疫细胞功能。 最近的临床前试验已经证明,在多种不同的动物模型中NAD+耗竭均是年龄相关疾病(包括神经退行性疾病、代谢性疾病和早老症)的关键途径。 但是,尚不清楚在特定疾病中哪些细胞和酶会导致NAD+水平下降,哪些靶标或途径可用于有效和安全地恢复NAD+动态平衡仍在研究中。 但是已证实多种不同的NAD+增强策略可有效延长健康期和寿命(请参阅“NAD+水平下降的治疗靶点”部分和表 1)。 因此,使用NAD+前体(例如NMN和NR),以及促进NAD+生物合成或抑制NAD+降解的小分子,为治疗与衰老相关的疾病和增加人类健康水平提供了令人兴奋的治疗方法。 这一点尤其重要,因为老年人口正在迅速增加,并且衰老相关疾病预计将在未来几十年内给社会造成沉重负担。 然而,NAD+增强疗法能否真正转化到临床为人类所用仍然是需要回答的关键问题。目前一些人类临床试验正在进行,以评估NAD+增强的安全性和有效性(表 2),短期NR / NMN给药的早期试验已证明它是安全的,并且可以提高健康参与者的NAD+水平。 然而,尽管初步结果令人鼓舞,但长期补充NAD+前体是否有任何副作用仍是未知的。 此外,还需要回答许多其他问题,以加深人们对NAD+增强疗法潜力的认识:NMN和NR是否存在任何组织/疾病特异性? 对于不同疾病,NAD+前体的治疗剂量是多少? 是否可以考虑将增强NAD+的策略(例如CD38和/或PARP1抑制剂)与补充NAD+的前体相结合? 希望目前进行中的临床试验能够在不久的将来为这些尚未解决的问题提供一些启示,并为未来的发展方向奠定基础,以解读NAD+在人类衰老过程中的作用。

来源:老顽童说
原标题:《【重磅综述】机体衰老与NAD+之间不得不说的故事》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2024 上海东方报业有限公司

