- +1
急急如律令,血拼急性早幼粒细胞白血病四部曲
原创 高晓鹏 黄兴琴 检验医学网
作者:高晓鹏[1]、黄兴琴[2]
单位:1.西安市儿童医院中心实验室
2.重庆市陆军军医大学西南血液科实验室
导读
关于急性白血病,目前形态分类已经不是最终诊断,如果可以简化到AML, APL, ALL 等大框框诊断,那么APL(急性早幼粒细胞白血病)始终是诊断的重中之重,诊断必须快上加快,早一天诊断,早一天治疗,结局完全不一样,阴阳之隔一线间。坚守平日江苏省人民医院张建富老师对我们形态工作者病例教学的简单和多次强化原则,关于APL我们从四大部分进行总结,包括:临床表现、形态特征、免疫分型、遗传和分子生物学(MICM)。
APL临床曲
APL四部曲(合集之一)
APL的发病率占同期AML患者的10-15 %,在高龄患者中的占比要低些。任何年龄均可发病,但大部分患者为中青年人,平均发病年龄为44岁,发病率约为0.23/10万。为了强调融合基因PML-RARa的意义,WHO 2016分型将APL归类为AML伴重现性遗传学异常,WHO造血和淋巴组织肿瘤分类2017年修订为APL伴PML-RARa。
01
APL除具有一般AML的共同临床特征即有发热、贫血和出血外,APL临床表现中出血(往往是较严重出血)是其特征性表现,出血以皮肤黏膜为主,比如牙龈肿胀渗血、口腔粘膜血泡;其次为消化道、泌尿道、呼吸道、颅内出血,比如胃肠道出血、肺出血、非妇科疾病明显的阴道出血,育龄女性可能会月经过多,致命的是中枢出血。APL肝脾淋巴结多数无肿大。但是初诊时50%以上的病例无出血症状,一旦出血进展迅速;起病及诱导治疗过程中容易发生出血和栓塞而引起死亡。

出血、出血、出血
BLOOD、BLOOD、BLOOD!
重要的事情说三遍!
02
APL外周血常规绝大多数为三系减低,可高达80%,但也有约20%患者白细胞总数正常和增高等。以FAB分型的M3a多见白细胞减少,甚至全血细胞减少,贫血和血小板减少多为严重,主要原因是异常增殖的白血病细胞排挤了红细胞和血小板的生成,血小板减少更为明显;M3b和M3v多见白细胞增多,M3v (即微颗粒型 APL)白细胞数非常高,倍增时间快,并可伴有脾大。但是粗颗粒变异型APL可能外周血白细胞总数增高的。其实不管什么病案,绝大多数都是临床和诊断符合的,但是总有不按常理出牌的。APL散点图可见异常早幼粒细胞弥漫分布于单核/幼稚粒细胞群之间,呈泪滴状散点图。
03
有明显的凝血障碍,凝血指标比如APTT、PT、Fg和DD二聚体异常,有低纤维蛋白血症,D-D二聚体增高等,提示有出血倾向,易发生DIC。APL一般都伴有比其他类型AML更加严重的出血症状,出血因素与血小板减少固然有关,但最主要因素是异常早幼粒细胞胞质中颗粒释放促凝物质和蛋白水解酶,引起DIC和纤溶亢进,发生广泛而严重的出血,导致APL病情凶险。
04
高凝骨髓。病人往往有骨髓浸润症状(胸骨压疼等),提示医生骨穿需要注意髓液有无很快出现高凝现象,往往刚刚从针管打在玻片上就发生凝集,呈现推不开的骨髓,必须快速制片,有经验的老师可以快速推出3-5张片子,液片往往呈倒三角的稠密状态,多数能推出两三张骨髓液就凝集呈块了,此时尽量蘸取周围髓液多制片。操作时遇到这样的情况要想到APL或者M5(出血或者脾大的往往也是凶险),尽快染色尽快镜下看,迅速与临床沟通告知形态学倾向,医生可根据病情做出相关预处理治疗,我们形态人的争分夺秒就是帮助患者和死神赛跑!
05
APL药物致病史。目前报道较多的是牛皮癣用乙双吗啉而导致的APL。临床部分治疗牛皮癣药物比如:乙亚胺、双酮嗪、乙双吗啉等药物均可致染色体畸变,多在服药2-7年中发生APL,上述药物因致白血病作用已停产;肺癌患者使用吉非替尼药物,可能也导致APL,在肺癌治疗过程中的表皮细胞生长因子抑制剂与APL发病相关。APL绝大多数患者为原发性,据报道有10%左右APL是治疗相关的,对于牛皮癣和肺癌患者的用药史要问询清楚。
06
APL治疗用药。APL被认为是通过药物,不需要骨髓移植就可治愈的唯一的AML;目前多采用王振义院士和陈竺院士提出的上海方案,即以维A酸和三氧化二砷(As2O3)为基础的双诱导或辅以化疗的三诱导方案治疗。ATRA(全反式维甲酸)目的是推动异常早幼粒向成熟分化,让早幼粒细胞不破损,防止细胞释放的颗粒加速凝血功能障碍致严重出血,避免致命的出血并发症。砷剂的作用使得异常克隆转为正常克隆,可使RARa转阴。难治和复发患者对三氧化二砷反应良好,因此大部分患者可以长期存活,甚至治愈,但伴有CD56表达者预后相对较差!若查见异常早幼粒,怀疑患者APL,应作为危急值来报告临床,而临床医生对疑为APL的患者,也常会立即使用全反式维甲酸,尽早给予ATRA治疗是防治凝血功能异常最重要的因素,随后根据融合基因的检测结果调整用药。对于高白APL单用ATRA诱导分化则会加重负荷,会更危险,应加ATO诱导细胞凋亡,进行双诱导治疗。2019NCCN指南指出,当首次怀疑APL时启动ATRA治疗,若细胞遗传学和分子检测不能证实APL则停用ATRA,按照AML方案治疗。APL已成为基本不用进行造血干细胞移植即可治愈的AML,并成为转化医学的典范。
APL形态曲
APL四部曲(合集之二)

APL的典型细胞具有四大特征:
内外浆、不规则核形(蝶型核、臀样核),柴捆棒子(Auer,S)、POX强阳性。
FAB将M3型分为3型:
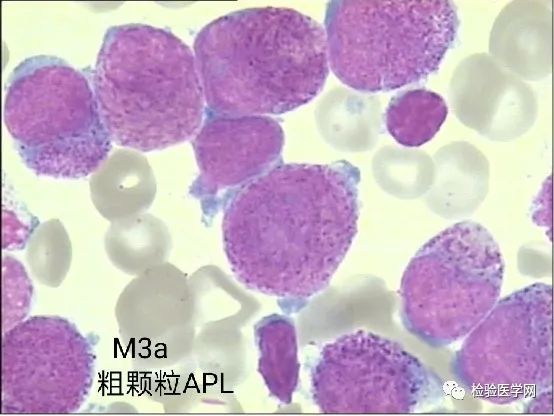
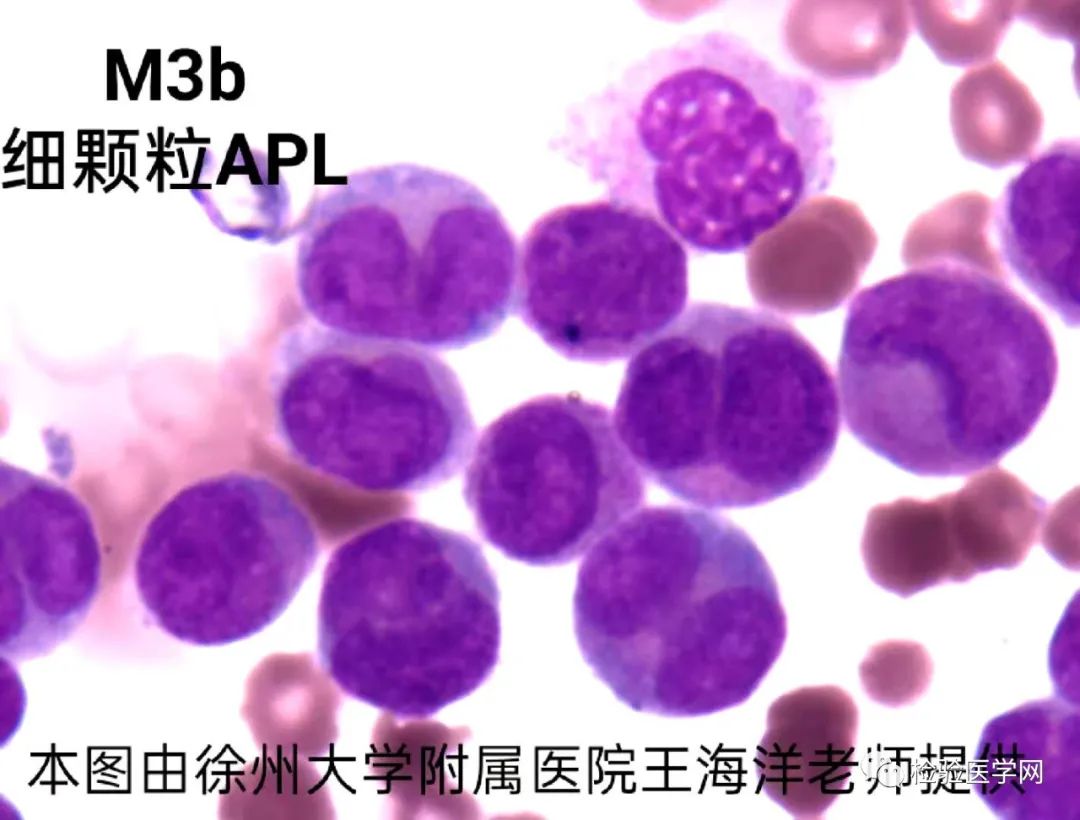
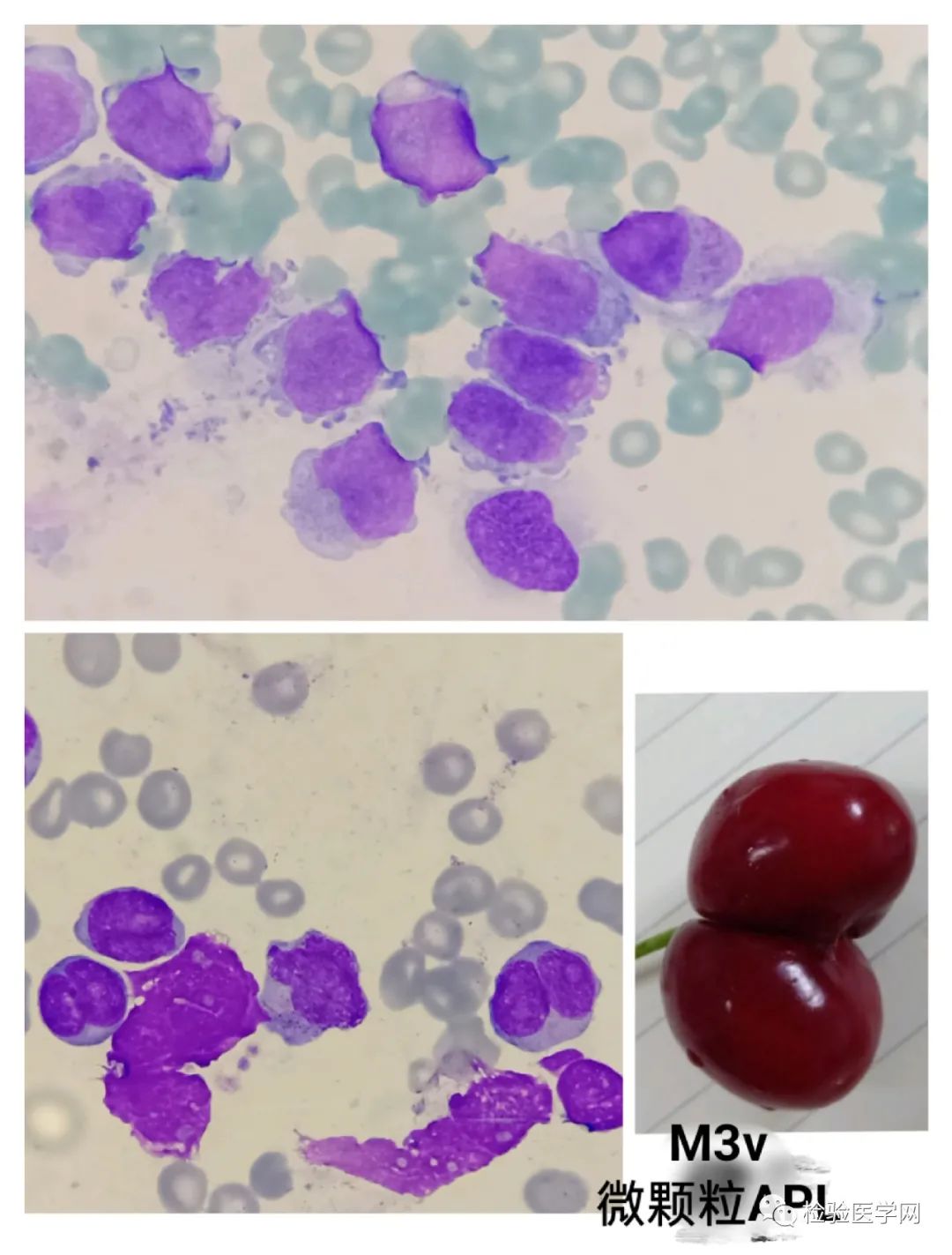
M3a(胞质粗颗粒型)、M3b(胞质细颗粒型)、M3v(胞质微颗粒型,颗粒甚少或缺如,核型扭曲、分叶)。
(点击图片可放大)
WHO强调了重现性遗传学遗传的重要性,专门将APL伴PML-RARa列为一类。M3和APL不完全等同,前者以形态学为主,后者强调了融合基因PML-RARa。APL看异常早幼粒,强调的一定是异常两字,即使正常早幼粒形态比例>50%都不能诊断APL,在粒细胞缺乏症或者白血病化疗过程中用了刺激因子后早幼粒细胞比例都可能明显增高,胞体大颗粒粗且松散局部密集,核往往大而规则。假如是异常早幼粒细胞,胞体大小不一,倒三角或降落伞样,胞质突起,核形变异,颗粒粗细不一,即使全片比例不足20%,也要提示临床APL可能性大,建议结合染色体和融合基因检查。
01
内外浆:异常早幼粒细胞大小不一,胞质丰富,可见内外胞质现象,胞质边缘为外浆染蓝色呈伪足样突起,胞质内由于颗粒影响往往偏红,内浆有的充满密集的紫红色嗜天青颗粒(粗颗粒),颗粒粗大深染密集融合常常遮盖于核上;有的则为细颗粒或微小颗粒如灰尘样肉眼不易见。紫红色嗜天青颗粒(又称联苯胺蓝颗粒)是促凝颗粒,引起DIC主要原因之一。M3v其早幼粒细胞高核质比,胞浆颗粒少或无,甲苯胺蓝染色阳性。
02
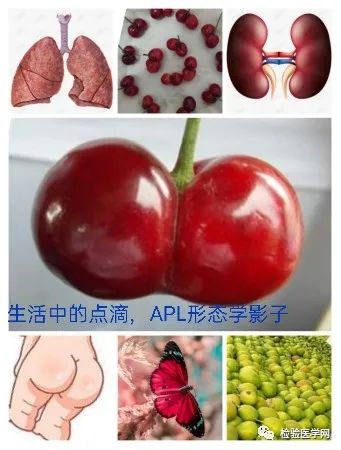
不规则核形,也是异常早幼粒细胞的特征性表现。常呈大小核、肾形、双叶形、双核、类臀样、类蝴蝶样、香梨样、连体樱桃样等,似单核样核;核染色质致密,有的可见模糊核仁。个人体会M3a胞质粗颗粒多且积聚于一侧,核形蝴蝶翅膀翩翩起舞样,整个细胞犹如降落伞或花苞,M3b、M3v颗粒细小或无颗粒,核形不规则特征突出,更象双胞胎连体樱桃或者车厘子模样。其实不管形态像什么,只要我们能根据个人经验的积累识别出异常才是关键。
03
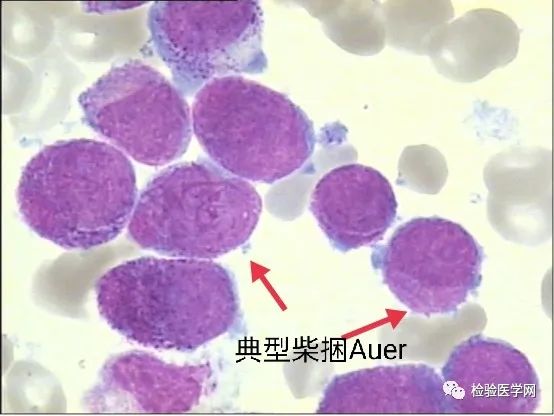
柴捆Auer
M3a易见单个和/或柴捆状Auer,S小体,即柴捆(faggot)细胞,这是诊断急性早幼粒细胞白血病的有力证据;M3b可见或偶见Auer小体,M3v一般不见Auer小体,此亚型与M5细胞形态很难鉴别,但做了POX后很好鉴别,M3v呈强阳性,原幼单MPO多为弱阳性或阴性。再者幼单胞浆不像是内外浆现象,仅是有时伪足突起,浆染色不匀。柴捆状Auer,S为多条细长的柴棒,束状交叉排列,如同剑池,如同乱柴,有的细胞颗粒不典型,但是见到多条Auer小体者可帮助明确诊断。
04
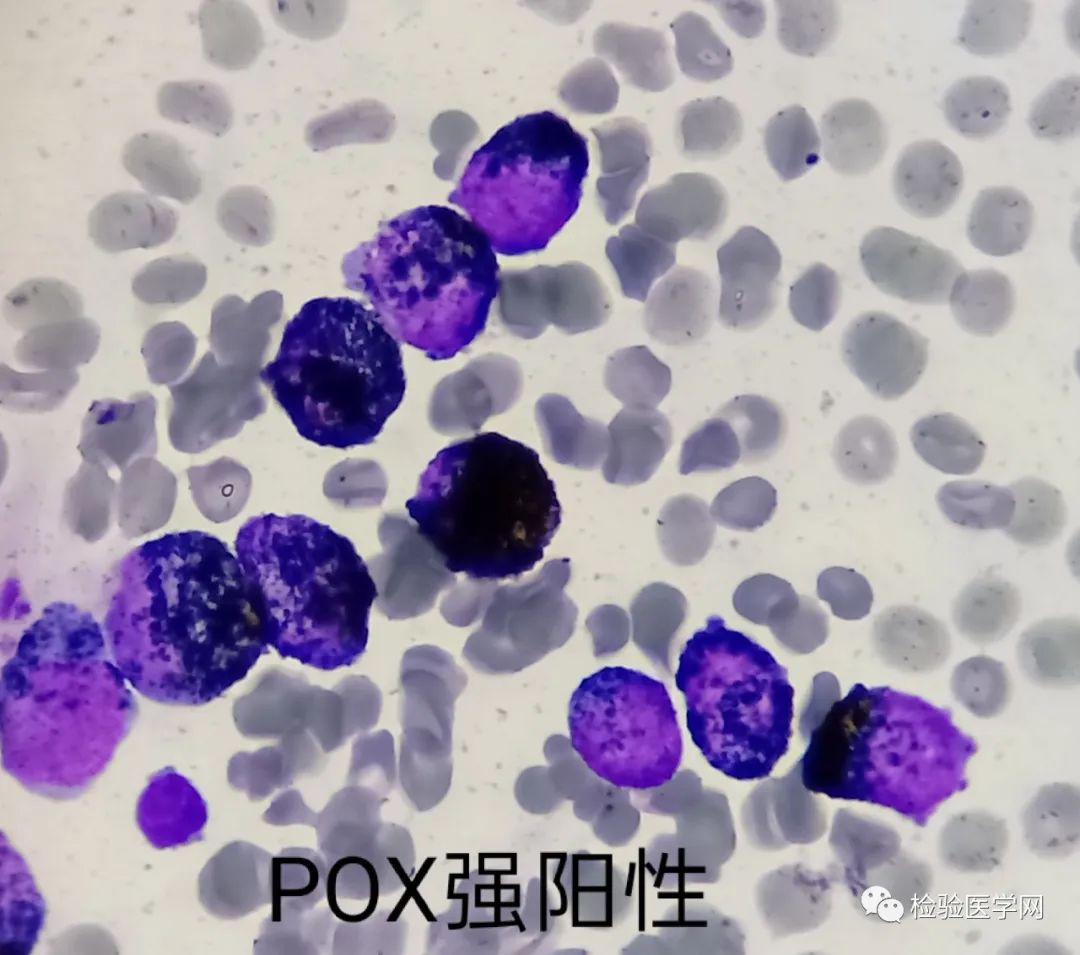
细胞化学染色,异常早幼粒不论有无颗粒POX多呈强阳性反应,极个别可呈弱阳性或阴性,SBB和CE也多呈强阳性,往往在POX或者CE染色中可见胞浆针状结晶,根据我们的经验就是Auer小体。PAS异常早幼粒多为弱阳性反应,即胞质呈粉红色。
形态诊断中的注意事项
01
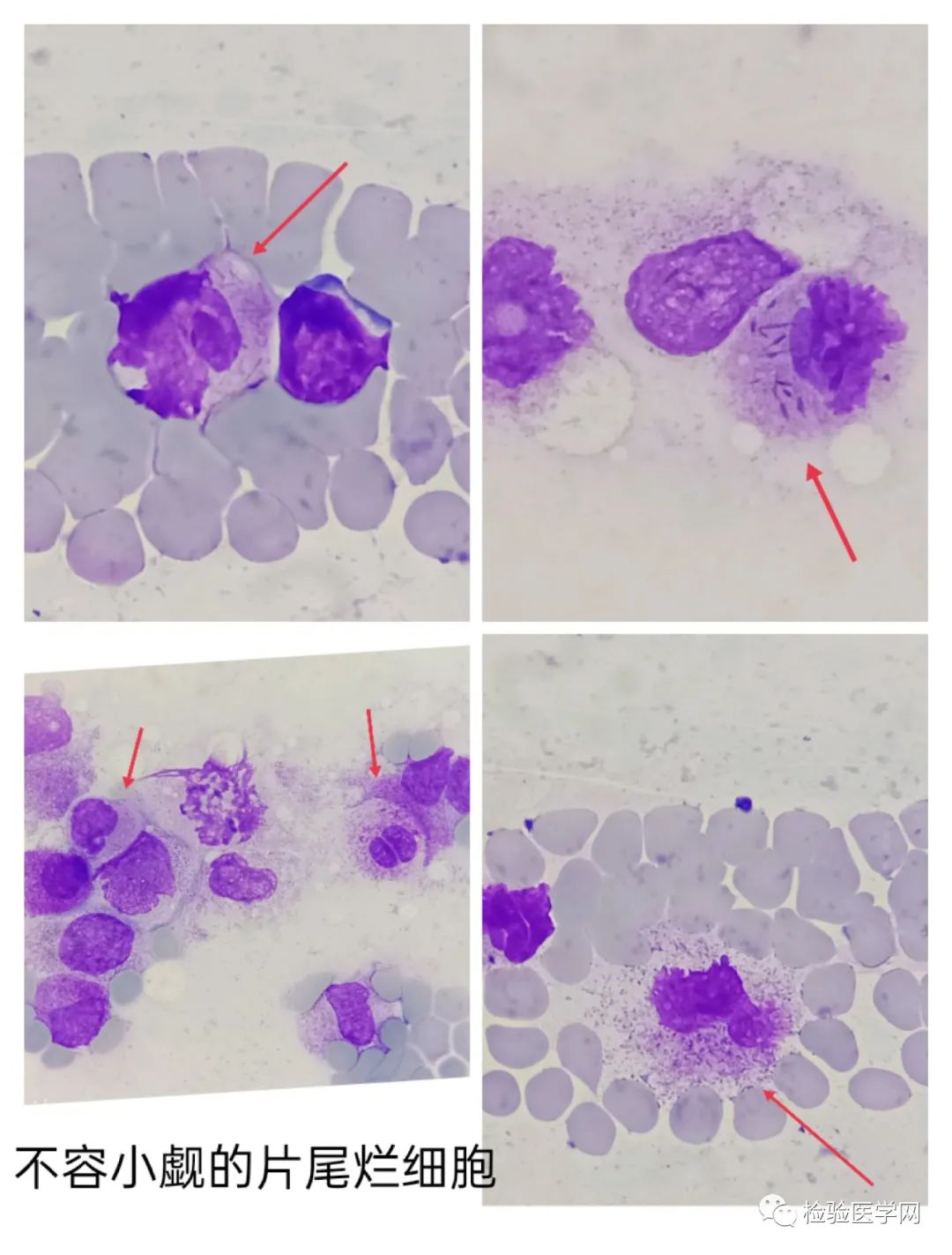
外周血细胞形态分析,对于起病急且全血细胞减少的出血患者行外周血细胞形态分析时,应疑及APL。对白细胞低的必须全片浏览,要注意涂片的边缘与尾部(即海岸线)的类单核样细胞和/或典型的颗粒过多的早幼粒细胞,特别是破碎细胞胞质中易见奥尔小体,注意柴捆细胞。要阅片观察,不仅仅分类,宁可错杀,不可放过,并及时和临床沟通。对于白细胞总数增高患者,细胞形态类似单核时要想到APL,此时要多部位观察看有无柴捆细胞。(张建富老师多次病例讲解强调的!很实用!)
02
骨髓象
多增生明显活跃或极度活跃,红、巨两系受抑,异常早幼粒细胞占比达85%以上,截头去尾现象,原始粒细胞比例多<5%,即原始不能多,中后不能多。原始细胞增多可能是颗粒增多的M2或M4;中幼以后增多可能是伴病态的M2或AML-MRC。粗颗粒异常早幼粒细胞很好识别,但是细颗粒或微小颗粒的需要和M5和M2b鉴别,后两者缺乏典型的粗颗粒早幼粒细胞以及典型的核形或柴捆小体,原始细胞比例往往>10%;APL往往一张片子里粗细颗粒都可见,内外浆明显。见柴捆细胞基本可以肯定为急性早幼粒细胞白血病,但是也不一定,也有报道不少AML可见柴捆Auer,S小体,但是并非APL;在APL病例中,也有类似包涵体样物质,其本质是嗜天青颗粒融合形成的“圆奥”小体,此时要注意与其他类型白血病的鉴别,特别是M5。POX染色是较好的鉴别实验,马上行POX染色,强阳性者为M3,弱阳性或阴性者可能是M5;NBE染色M5阳性,APL阴性。
03
与急性嗜碱性粒细胞白血病鉴别:嗜碱颗粒少量、散在、粗大,覆盖整个核,更易见成熟阶段的嗜碱性粒细胞。甲苯胺蓝染色阳性。APL伴有嗜碱性粒细胞增多的病例少见,临床表现多见粘膜出血。
04
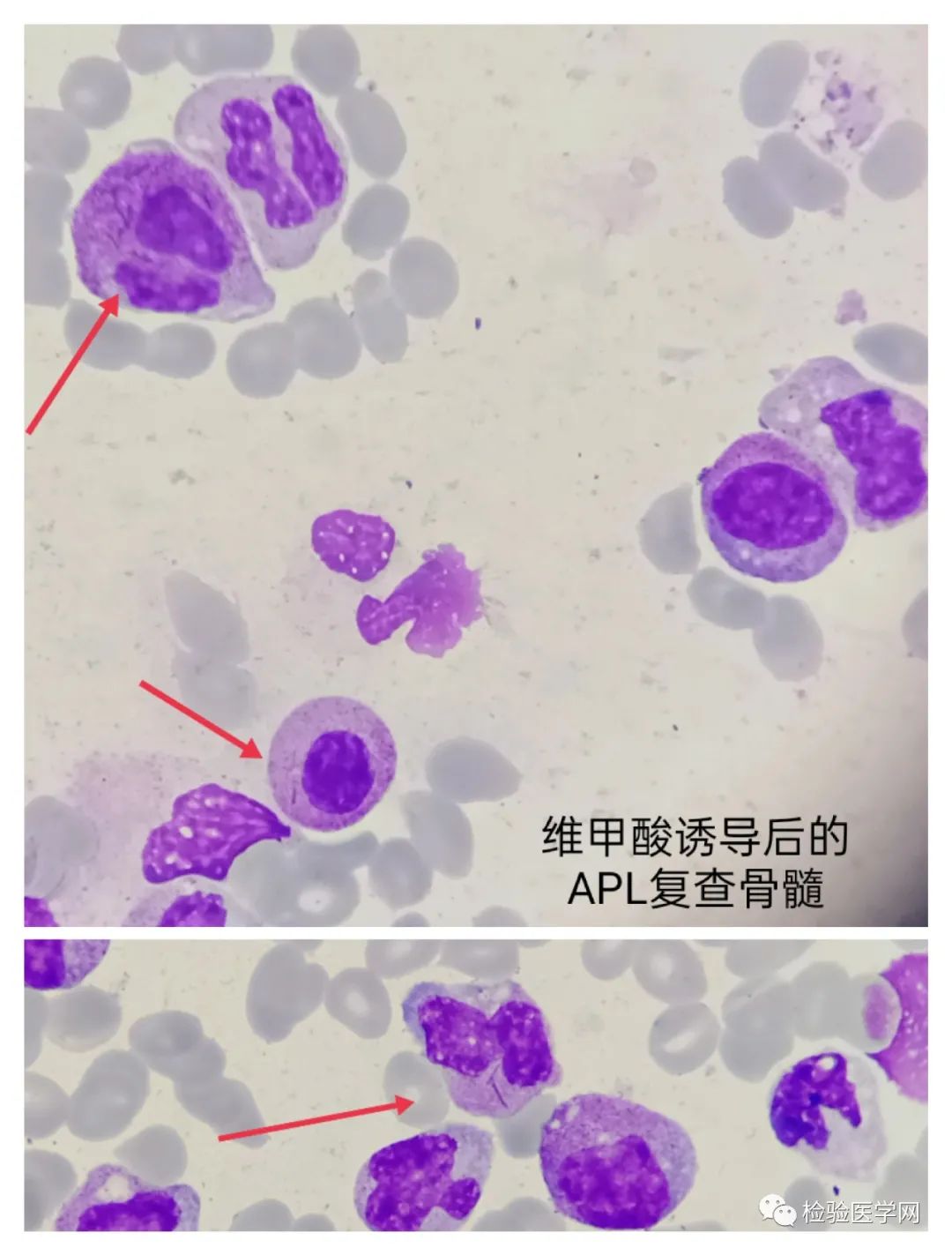
APL经过ATRA诱导分化治疗后的复查骨髓像,见到较多的往下分化的下阶段细胞,部分细胞可见内外浆,核扭曲、折叠、双分叶、蝴蝶形,下阶段的中晚杆分均可见,胞浆中见单根和多根Auer,S小体,此为APL未缓解阶段;有些细胞胞浆中颗粒减少,胞浆粉粉的,无较多嗜天青颗粒,这是APL在治疗过程中的脱颗粒现象,与初诊形态不一致。
APL免疫分型曲
APL四部曲(合集之三)
典型的APL免疫分型:
流式SSC大、干细胞抗原CD34和HLA-DR不表达或低表达,CD117阳性,髓系分化抗原强表达MPO、CD33、CD15、CD13等,粒系分化标志 CD15 和 CD65 为阴性或弱阳性,但常表达 CD64,共表达CD2和CD9。免疫表型不是APL确诊的依据,分子学证据依靠FISH和RT-PCR。
在APL中,只有75%的病例存在CD34-HLA-DR-、CD117+的表型,所以为了更有特异性的诊断APL,需要加入CD11b和CD11c这两个粘附分子(CD11b-CD11c-),就可以形成一组对APL有100%特异性的指标。如果流式DR/34阴性,11b/c阳性,可理解为一定程度下往中幼阶段分化了,肿瘤细胞发育并不能完全套正常细胞的发育模式,早晚标记会出现错乱表达。少数形态学为微颗粒型或者少颗粒型或者PML-RARa融合基因为bcr3转录本的病例,SSC偏小,常表达CD2、CD34、HLA-DR(可部分表达),有的CD34和HLA—D R强表达,部分细胞还可表达CD11c,CD2在APL的表达通常与FLT3-ITD有关,FLT3-ITD突变最常见与高白、细颗粒型原始细胞形态和累及PML的bcr3断裂点相关,而前述的免疫表型均缺失,此时分子学往往可发现S型PML-RARa。约 10% 的 APL伴CD56表达,往往提示预后差。
APL遗传分子曲
APL四部曲(合集之四)
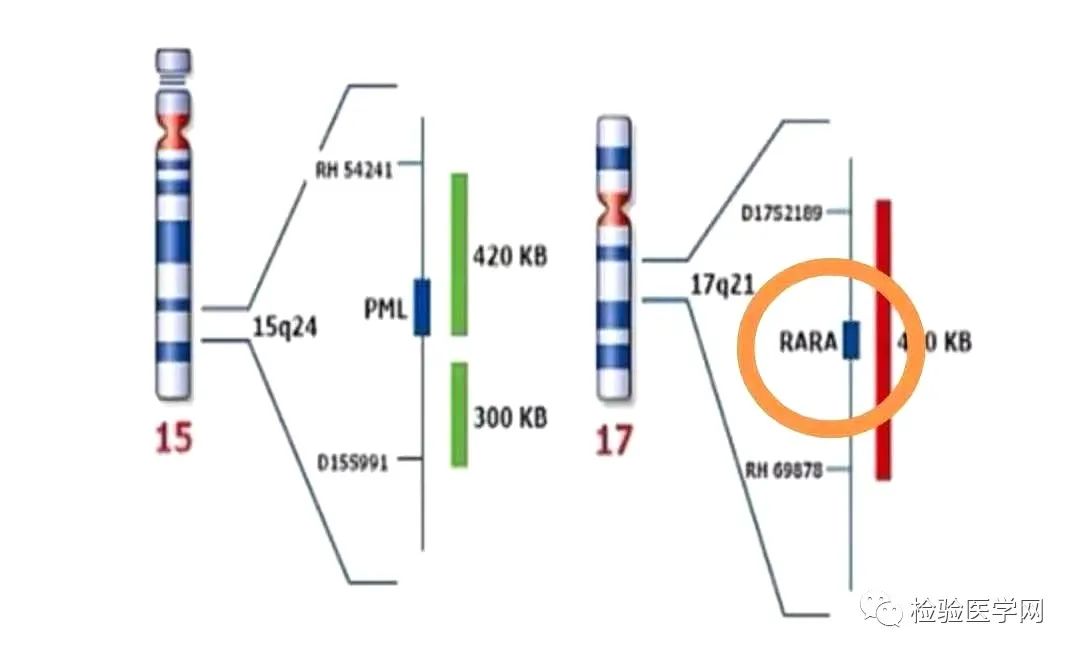
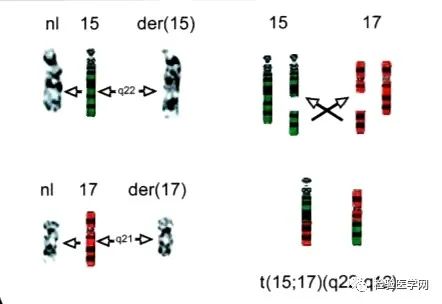
染色体检查及分子生物检查,诊断APL的金标准是RARa阳性。快速诊断可以用FISH方法和Q-PCR。染色体核型常见t(15;17)(q24;q12)和PML-RARa融合基因阳性。PML-RARa融合除了见于t(15;17)(q24.1;q21.2)易位外,还可以是隐蔽易位或复杂的细胞遗传学重排导致,为了强调该融合基因PML-RARa的意义,WHO 2017分型中修订为APL伴PML-RARa,该类白血病没有强调染色体,仅强调基因阳性就能诊断。98%的APL患者存在PML/RARa融合基因,另有低于2%的APL患者为其他类型融合基因,精确的分子生物学诊断可为临床选取治疗方案提供依据。
01
常见t(15;17)(q24;q21)和PML/RARa约占98.0%,PML/RARa对APL诊断具有特异性,也是治疗的分子生物学基础,所以WHO 2017分型中修订为APL伴PML-RARa,就是提高了融合基因的地位。t(15;17)染色体易位,使17号染色体上的维甲酸受体RARa基因发生断裂,与15号染色体上的早幼粒细胞白血病基因PML发生融合,形成PML/RARa融合基因,该基因的蛋白产物导致细胞分化阻滞和凋亡不足,是APL发生的主要分子机制。本型G带形态特点为1条15号染色体长臂远端出现深染带,似乎有3条14号染色体,而17号染色体中的1条长臂均为阴性带。从基因水平PML/RARa具有三种类型BCR1、BCR2、BCR3,也称为长型(L)、变异型(V)和短型(S),BCR1型幼稚细胞为典型的多颗粒型,胞体大,胞浆丰富,可见内外浆及粗大颗粒,核形不规则,核染色质细致,核仁可见,可见Auer小体,免疫表型为典型APL特点。BCR2型无明显特殊性。BCR3型幼稚细胞常为微颗粒型,FCM可表现为SSC值比典型APL低,部分患者表达CD34,但多数患者免疫表型和BCR1型无明显分别。本类型对全反式维甲酸敏感,生存期长。
(点击图片可放大)
02
还有APL伴变异型RARa易位,尽管WHO分类不算伴重现性遗传学异常的AML,也是APL,涉及到隐匿异位、插入、复杂重排。2020版CSCO恶性血液病诊疗指南发布变异型APL近13种之多。
如:t(11;17)(q23;q12)和PLZF/RARa(锌指基因),现在叫ZBTB16-RARa约占0.8%,在WHO2017版则更新为APL伴变异型RARa易位(见WHO蓝皮书P136)。临床中十分少见,大约占APL不到1%的比例,形态像多颗粒M2,形态学特征即为异常细胞核形规则,颗粒多,常无Auer,S小体,Pelger样的中性粒细胞增多,MPO强阳性,易误诊为多颗粒AML-M1/2,M2b。对维甲酸治疗不敏感,单独应用维甲酸诱导分化治疗,疗效欠佳,有报道采用维甲酸联合化疗、或亚砷酸联合化疗的方法可达CR,但长期效果欠佳。
03
t(5;17)(q35; q12)和NPM/RARa(5q35上核磷蛋白基因),初期的伴有t (5; 17)APL病例以多颗粒早幼粒细胞为主,有少数少颗粒早幼粒细胞,常无Auer,S小体,对全反式维甲酸有反应。
04
t(11;17)(q13; q21)和NUMA1/RARa(核基质相关基因)。对全反式维甲酸敏感。
05
der(17)dup(17)(q12; q12)和STAT5b/RARa,对全反式维甲酸无效。der(17)t(17;17)(q24; q12)PRKAR1a/RARa对全反式维甲酸有反应。
06
t(4;17)(q12; q21)和FIP1LI/RARa,少见类型。本型细胞胞质内外浆明显,颗粒粗大,核形较规则类似被造血因子刺激后的早幼粒形态,颗粒更大似桑葚的表观或者刺猬模样。对全反式维甲酸有反应。
07
t(X;17)(p11; q21)和BCOR/RARa对全反式维甲酸有反应。
08
t(7;17)(q11; q21)和GTF2I/RARa对全反式维甲酸无效。
09
t(1;17)(q42; q21)IRF2BP2/RARa对全反式维甲酸有反应。
10
还有t(2;17)(q32; q21)OBFC2A/RARa、t(3;17)(q26; q21)TBLR1/RARa、t(17;17)(q21; q21)STAT3/RARa、t(1;17)(q42; q21)FNDC3B/RARa等变异型。
11
极为罕见的i(17)(q10)类APL的AML,其形态学和M3a极为相似,但是染色体异常是由于17号染色体着丝点发生损伤或不合理的断裂,导致其短臂缺失,长臂发生等臂而形成。. FISH检测:nuc ish(PML×2,RARA×3),PML/RARA易位探针未见融合信号,RARa(位于17q21)位点拷贝数增加。用全反式维甲酸及三氧化二砷无效,用AML方案强烈化疗不佳,有报道单独i(17)(q10)染色体异常疗效差。该类型RARa拷贝增加的,形态学类似APL的,是否也是APL伴变异型RARa易位呢?还需收集病例进行分析研究。
12
部分病例可见继发性细胞遗传学异常,最常见的是+8染色体异常(10%~15%)和FLT3-ITD(34%~45% )突变,包括内部串联重复(ITD)和酪氨酸激酶域(TKD)突变。FLT3- ITD 突变最常见与较高的白细胞计数、低血小板有关,细颗粒型原始细胞形态和累及 PML 的bcr3 断裂点相关。既往曾认为伴有FLT3-ITD基因突变的APL预后较差或容易复发,但2019年ELN研究提示:FLT3-ITD基因突变并不影响诱导治疗效果及APL预后。
总结
APL的诊断能否及时准确发出,是细胞形态学工作者的重中之重,临床特征早诊断,早治疗,是本型的特征。牢抓APL形态学四个特征:内外浆、不规则核形、柴捆棒子、POX强阳性,具备其中1-2个形态特点就需要考虑异常早幼粒细胞。确诊APL的金标准是RARa融合基因阳性。除个别基因类型疗效较差外,绝大多数患者可以长期存活,可以说是治愈。作为细胞形态学工作者,我们必须不断修炼自己,积累细胞形态经验,让我们拥有一双慧眼,让患者受益。
特别致谢江苏省人民医院血液科实验室张建富教授的点滴教导!致谢王海洋老师提供图片。作者学识有限,难免有不足和错误之处,配图为网络搜图或确诊病例手机采集,清晰度不足,恳请专家和老师们提出批评指正!
原标题:《急急如律令,血拼急性早幼粒细胞白血病四部曲》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2024 上海东方报业有限公司

